Exhibit 99.1
Business
Except as otherwise specified or indicated by the context, references to “we,” “us,” “our” or the “company” are to TuHURA Biosciences, Inc. and its wholly owned subsidiaries.
Overview
We are a clinical stage immuno-oncology company with three distinct technologies focused on the development of novel therapeutics designed to overcome primary and acquired resistance to cancer immunotherapies.
Our proprietary Immune FxTM technology platform, or IFx, is an innate immune agonist technology designed to “trick” the body’s immune system to attack tumor cells by making tumor cells look like bacteria. Our lead product candidate, IFx2.0, is an innate immune agonist designed to overcome primary resistance to checkpoint inhibitors. In June 2025, we initiated a single randomized placebo-controlled Phase 3 registration trial of IFx-2.0 administered as an adjunctive therapy to Keytruda® (pembrolizumab) in first line treatment for patients with advanced or metastatic Merkel cell carcinoma who are checkpoint inhibitor naïve utilizing the FDA’s accelerated approval pathway.
In addition to our IFx technology platform, in June 2025 we acquired the rights to TBS-2025, a novel VISTA-inhibiting monoclonal antibody formerly known as KVA1213, through our acquisition of Kineta, Inc. (“Kineta”) on June 30, 2025. VISTA (otherwise referred to as V-domain Ig suppressor of T cell activation) is an immune checkpoint highly expressed on myeloid cells that is believed to be a strong driver of immunosuppression in the tumor microenvironment and is believed to be a primary mechanism by which leukemic blasts escape immune recognition contributing to low relapse rates and high rates of recurrence in acute myeloid leukemia, or AML. Following our acquisition of Kineta, we are currently planning on investigating TBS-2025 in a randomized Phase 2 trial in combination with a menin inhibitor vs menin inhibitor alone in mutated NPM1 (mutNPM1) AML.
In addition to our IFx and TBS-2025 technologies, we are leveraging our Delta Opioid Receptor technology to develop tumor microenvironment modulators in the form of first-in-class bi-specific antibody-peptide conjugates (“APCs”) and antibody-drug conjugates (“ADCs”) targeting Myeloid Derived Suppressor Cells (“MDSCs”). Our APCs and ADCs are being developed to inhibit the immune-suppressing effects of MDSCs on the tumor microenvironment to prevent T cell exhaustion and acquired resistance to checkpoint inhibitors and cellular therapies.
IFx Innate Immune Agonist Development Program
We have developed Immune FxTM, or IFx, as an innate immune agonist technology designed to “trick” the body’s immune system to attack tumor cells by making tumor cells look like bacteria and to thereby harness the natural power of innate immunity by leveraging natural mechanisms conserved throughout evolution to recognize threats from foreign pathogens like bacteria or viruses. Our innate immune agonist product candidates are delivered either via intratumoral injection (in the case of the Company’s pDNA innate immune agonist) or tumor targeted via intravenous or autologous whole-cell administration (in the case of our mRNA innate immune agonist).
Our IFx-2.0 innate immune agonist, our company’s lead product candidate, is comparatively simple to administer and involves only the injection into a patient’s tumor, or lymph node, of a relatively small amount of pDNA that is designed to encode for an immunogenic gram positive bacterial protein that gets expressed on the surface of the patient’s tumor so that the surface of the tumor looks like a bacterium.
Bacteria, like all pathogens, have molecular patterns or motifs that are conserved through evolution and that are recognized by specific pattern-recognition receptors on immune cells of our innate immune system. This is an individual’s primary line of defense against pathogens that the individual is born with, and the innate immune system has no choice but to recognize the tumor as it would a gram-positive bacteria or any pathogen. Gram-positive bacterial proteins are recognized by Toll Like Receptor-2 (TLR-2) on antigen presenting cells, or APCs, which engulf and ingest the entire intact tumor cell packaging all the foreign tumor neoantigens presenting them to and educating tumor killing T cells and B cells. In doing so, IFx-2.0 harnesses the power of the innate immune response to produce activated tumor-specific T cells where they previously didn’t exist overcoming primary resistance to checkpoint inhibitor therapy.
We have entered into a Special Protocol Assessment agreement with the FDA for a single Phase 3 randomized placebo and injection-controlled trial for IFx-2.0, our lead innate immune agonist, as an adjunctive therapy to pembrolizumab (Keytruda®) in the first line treatment of patients with advanced or metastatic Merkel cell carcinoma, who are checkpoint inhibitor-naïve utilizing the FDA’s accelerated approval pathway. A Special Protocol Assessment agreement is a binding written agreement between the U.S. Food and Drug Administration (FDA) and a trial sponsor that indicates the FDA has agreed to the study’s design, charters, and statistical analysis plan, and if the study endpoints are met within the context of the SPA Agreement, such results would be adequate to support accelerated and regular approval. A Special Protocol Assessment agreement does not increase the likelihood of marketing approval for the product and may not lead to a faster or less costly development, review, or approval process. We initiated the Phase 3 trial in June 2025.
In designing the Phase 3 trial for IFx-2.0, we worked with the deputy director of the FDA’s Oncology Center of Excellence (OCE) on what we believe is a unique trial design. Consistent with the FDA’s Project Front Runner initiative, the FDA recommended investigating IFx-2.0 in the front-line treatment setting rather than in patients who are progressing on checkpoint inhibitor therapy. In doing so, data from a primary endpoint of objective response rate, or ORR, that is of sufficient magnitude and duration and with a favorable risk/benefit profile could be sufficient to support accelerated approval. Furthermore, OCE requested that the Company consider incorporating a key secondary endpoint that is of clinical benefit such that results from a key secondary endpoint of progression-free survival, or PFS, that is adequately powered with statistical assumptions in the statistical analysis plan provided to the FDA, if achieved without a detrimental effect on overall survival, or OS, could be adequate to support conversion to regular approval satisfying the requirement for a confirmatory trial.
We anticipate that enrollment for the Phase 3 will take approximately 14 – 18 months from the initiation of the trial, with top-line data potentially being available 6 to 7 months following the last patient enrolled. If successful, this Phase 3 trial would form the basis of a Biologics License Application, or BLA.
We previously announced that we were pursuing development of a product candidate referred to as IFx-3.0, an mRNA innate immune agonist candidate for intravenous or autologous whole cell administration for blood- related cancers. However, with the acquisition of Kineta, we have determined not to advance the development of IFx-3.0 until the results of the IFx-2.0 Phase 3 trial in Merkel cell carcinoma are known and have reallocated resources to the below-described planned trial for TBS-2025.
TBS-2025 Development Program
As a result of our acquisition of Kineta in June 2025, we acquired the rights to TBS-2025, a novel VISTA- inhibiting monoclonal antibody formerly known as KVA1213. Unlike other checkpoints, which are mostly present on activated T cells, VISTA is predominately expressed on myeloid cells, notably MDSCs, and on quiescent T cells. Research has demonstrated that when mutated, NPM1 and DNM3TA, two of the most common mutations in AML and typically co-mutated in myelodysplasia (MDS), result in high expression of VISTA on the surface of leukemic blasts. The presence of VISTA on these cells is believed to be the primary mechanism by which leukemic cells escape immune recognition and attack, resulting in a low treatment response rate and a high level of relapse in AML.
TBS-2025 was previously investigated in a dose escalation Phase 1/2 trial, both as a monotherapy and in combination with pembrolizumab, in patients with relapsed and/or treatment-refractory advanced solid tumors.
TBS-2025 was well tolerated when administered every 2 weeks at doses up to 1,000mg both in the monotherapy arm (n=24) or in the pembrolizumab combination therapy arm (n=16). Pharmacokinetic and pharmacodynamic data demonstrated greater than 90% receptor occupancy across the every two- week dosing interval. Immunocytokine analysis was consistent with the mechanism of action for VISTA inhibition on immune cells.
We believe that, in a relatively inexpensive, small Phase 2 study, we can determine if TBS-2025 can augment the response rates seen with menin inhibitors and decrease the rate of relapse in patients with mutNMP1 relapsed or refractory AML where menin inhibitors are the current standard of care. Accordingly, we are currently planning on investigating TBS-2025 in a Phase 2 trial in combination with a menin inhibitor in mutNMP1 AML.
DOR Technology Development Program
In addition to its innate immune agonist product candidates, we are using proprietary Delta Opioid Receptor (DOR) technology to develop peptidomimetic or small molecule bi-specific/bi-functional immune modulating APCs and ADCs designed to inhibit the immune suppressing effects of tumor associated MDSCs on the tumor microenvironment to prevent T cell exhaustion and acquired resistance to checkpoint inhibitors. The Company’s DOR technology was developed by scientists at Moffitt Cancer Center and TuHURA Biopharma, Inc., a separate company whose intellectual property assets we acquired in January 2023 (“TuHURA Biopharma”) We believe the DOR represents a novel target to inhibit the immunosuppressive capacity of MDSCs through its control of the production of multiple immunosuppressive soluble factors, chemokines and direct cell-cell interactions.
The tumor microenvironment is the tissue surrounding a tumor, including the normal cells, blood vessels, and molecules that surround and feed a tumor cell and shield it from immune attack and eradication. MDSCs are a heterogeneous group of immature myeloid cells, which when recruited from the bone marrow to the tumor microenvironment, they transform to tumor-associated MDSCs which are characterized by their ability to suppress both innate and adaptive immune responses. Tumor associated MDSCs are generally believed to be a major contributor to T cell exhaustion (which is the loss of ability of T cells to proliferate and to kill cancer cells) and for acquired resistance to checkpoint inhibitors and cellular therapies like T cell therapies. The presence of tumor associated MDSCs in the tumor microenvironment or circulating in the bloodstream is highly correlated with poor prognosis and outcome in a wide variety of solid tumors and blood related cancers.
We believe we are the first company developing immune modulating APC/ADCs targeting the Delta Opioid Receptor on MDSCs. We are developing peptidomimetic or small molecule DOR-selective inhibitors to incorporate into our bi-specific, bi-functional APCs and ADCs, which we believe represents a paradigm shift from conventional APCs or ADCs that are currently in development or being marketed. Traditional ADCs are a class of drugs in which a monoclonal antibody is chemically linked to a cancer-fighting substance. The antibody carries the cancer fighting payload to the tumor cell improving the selectivity of the resulting anti-cancer activity. Next generation ADCs incorporate non-chemotherapeutic technologies to interfere with tumor cell cycle growth or to carry with the antibody a checkpoint inhibitor (so called “checkpoint ADCs”). In contrast, our APCs or ADCs do not target tumor associated receptor targets but rather target the Delta Opioid Receptor on MDSCs while carrying with them an immune effector to target a second receptor target like VISTA with a VISTA inhibiting antibody or other checkpoint inhibitor(s) producing novel bi-specific, bi-functional conjugates. These two functions are intended to work together with the goal of overcoming acquired resistance, preventing T cell exhaustion and allowing checkpoint inhibitors and cellular therapies to be safer and more effective while interfering with the tumor’s ability to invade and spread throughout the body.
Our Pipeline
Our pipeline focuses on acquiring and developing technologies designed to overcome tumor-intrinsic mechanisms underlying primary resistance to checkpoint inhibitors. We also focus on technologies to overcome acquired resistance to cancer immunotherapies related to the immune suppressing characteristics of the tumor microenvironment. We are leveraging our technology platforms to advance several diversified product candidates, including principally the following, each of which is described in more detail below.

Our History and Team
We were originally incorporated in Nevada on June 24, 2009 under the name Berry Only Inc. On January 25, 2013, TuHURA entered into and closed an exchange agreement, with Del Mar Pharmaceuticals (BC) Ltd. (“Del Mar (BC)”), 0959454 B.C. Ltd. (“Callco”), and 0959456 B.C. Ltd. (“Exchangeco”) and the security holders of Del Mar (BC). Upon completion of the exchange agreement, Del Mar (BC) became a wholly-owned subsidiary of TuHURA. On August 19, 2020, TuHURA completed its merger with Adgero Biopharmaceuticals Holdings, Inc., a Delaware corporation (“Adgero”), in which Adgero continued its existence under Delaware law and became a direct, wholly-owned subsidiary of TuHURA. Following the completion of the merger, TuHURA changed its name from Del Mar Pharmaceuticals, Inc. to Kintara Therapeutics, Inc. and began trading on Nasdaq under the symbol “KTRA.”
On October 18, 2024, TuHURA completed a reverse merger transaction contemplated by its Agreement and Plan of Merger, dated April 2, 2024 (the “TuHURA-Kintara Merger Agreement”), with TuHURA Biosciences, Inc., a Delaware corporation (“Legacy TuHURA”), and Kayak Mergeco, Inc., a Delaware corporation and wholly-owned subsidiary of TuHURA (“Kintara Merger Sub”). Pursuant to the TuHURA-Kintara Merger Agreement, Kintara Merger Sub merged with and into Legacy TuHURA with Legacy TuHURA surviving the merger (the “Kintara Merger”) and becoming TuHURA’s direct, wholly-owned subsidiary. TuHURA completed the merger on October 18, 2024, and changed its name on such date from Kintara Therapeutics, Inc. to “TuHURA Biosciences, Inc.” and begin trading on Nasdaq under the symbol “HURA.”
Legacy TuHURA’s predecessor company was formed as Morphogenesis, Inc. in 1995 by Drs. Patricia and Michael Lawman. Our IFx technology was developed in the laboratory of Dr. Michael Lawman at the Walt Disney Memorial Cancer Institute, where Dr. Michael Lawman was formerly a Director of the Institute, and Dr. Patricia Lawman was formerly Division Director of Cancer Molecular Biology at the Institute. Dr. Michael Lawman is a Fellow of the Royal Society of Biology, former Associate Professor at University of South Florida, and former Scientific Research Director of Pediatric Hematology/Oncology at St. Joseph’s Children’s Hospital. Dr. Patricia Lawman also serves as an Adjunct Professor at University of South Florida. Drs. Patricia and Michael Lawman are each inventors on numerous U.S. and foreign patents.
Our Delta Opioid Receptor APC and ADC technology was developed in the laboratory of Dr. Mark McLaughlin at the Moffitt Cancer Center and at the West Virginia University Research Corporation. Dr. McLaughlin was previously a Senior Member of the Drug Discovery Department at the Moffitt Cancer Center and previously Professor of Medicinal Chemistry and Member WVU Cancer Institute, where his research focused on protein-protein interaction inhibitor design and molecular targeted immunotherapy. The discovery that the Delta receptor is highly expressed on MDSCs was jointly discovered by scientists at Moffitt Cancer Center and TuHURA Biopharma, a separate company whose intellectual property assets we acquired in January 2023.
Our CEO, Dr. James Bianco, is a 33-year veteran of the biopharmaceutical industry. Dr. Bianco is the principal founder of CTI Biopharma, where he served as its CEO from 1992 to October 2016. Dr. Bianco’s experience spans all aspects of drug development from phase I-IV clinical trials, regulatory approval, and pricing reimbursement to sales and marketing. He has extensive experience in financing, negotiating and execution of pharmaceutical development and commercial license agreements. During his tenure at CTI Biopharma, Dr. Bianco was responsible for strategic portfolio development and identifying, acquiring, licensing, purchasing, or acquiring through international merger and acquisition, five drug candidates, four of which have since been approved by the FDA and with three receiving accelerated or conditional regulatory approval in the U.S. and/or E.U. In 2013, Dr. Bianco led CTI Biopharma in the identification and negotiation of the asset purchase for VONJO® (pacritinib), a novel JAK2 selective tyrosine kinase inhibitor. He also led CTI Biopharma in the negotiation of the development and commercial license agreement with Baxalta. As CEO of CTI Biopharma, Dr. Bianco was also responsible for the PERSIST-2 Phase 3 trial design and conduct, the successful results of which served as the basis for the 2022 FDA accelerated approval of Vonjo® (pacritinib) and the subsequent acquisition of CTI Biopharma by SOBI for $1.75 billion
Our Strategy
Our goal is to become a leading immuno-oncology company by developing novel therapeutics designed to overcome primary and acquired resistance to cancer immunotherapies, thereby broadening the impact of therapies such as checkpoint inhibitors. Our strategy is focused on leveraging our current technologies and novel product candidates and development programs in order to advance our current product candidates and expand our portfolio of products and technologies. The key elements of this strategy include:
| • | Shorten the time and cost to product registration. We are working to shorten the time and cost to product registration by focusing on patient populations that qualify for accelerated approval, such as patients with advanced and metastatic Merkel cell carcinoma in our Phase 3 trial for IFx-2.0. We believe this trial could significantly reduce the time and cost to potential approval and the cost associated with precluding the need for a postmarketing confirmatory trial. |
| • | Acquire and develop novel immunomodulatory technologies or product candidates targeting blood related cancers. Currently there are no cancer immunotherapies approved in blood-related cancers like AML or MDS, which presents an opportunity to develop novel agents to address such unmet medical needs. We believe we are uniquely positioned to identify, evaluate and potentially acquire novel drug candidates that focus on blood-related cancers that provide a strategic fit within our product pipeline and or with our DOR technology platforms. Our acquisition of TBS-2025 is consistent with this acquisition strategy and also provides synergy with our DOR technology providing the antibody for our ADC program. |
| • | Parallel development of differentiated drug product candidates within a therapeutic strategic focus on diseases with unmet medical needs like blood-related cancers. We believe a development program leveraging distinct technologies across a pipeline of differentiated drug candidates offers an efficient model of how small biotech companies can align capital and clinical development execution while managing technology and regulatory risks. We will continue to be opportunistic in acquiring drug candidates that are within our therapeutic strategic focus, like our recent acquisition of TBS-2025. In addition to providing a Phase 2 ready candidate to advance to clinical studies in mutNPM1 AML, we are investigating TBS-2025 when conjugated to a DOR inhibitor as our lead APC or ADC candidate in preclinical development. |
| • | Establish a leadership position in developing immune modulating bi-functional, bi-specific APCs and ADCs. We believe that we may be the first company to identify that the Delta Opioid Receptor is highly expressed on tumor-associated MDSCs and that it controls the regulation of multiple immune suppressive functions of MDSCs, the primary contributor to tumor microenvironment immunosuppression. We believe that inhibiting MDSC functionality may represent a novel way to overcome acquired resistance to immunotherapies. Our immune modulating bi-specific, bi-functional APCs and ADCs represent a paradigm shift in this important class of therapeutics and have the potential to position TuHURA to take the lead on advancing these novel immunomodulatory bi- specific, bi-functional ADCs and APCs to clinical trials. |
| • | Establish Development and Commercial License Collaborations. Leveraging our CEO’s track record of successfully establishing development and commercial partnerships with large multi-national pharmaceutical or biotechnology companies, we intend to seek and establish partnerships as a source of non-dilutive capital and funding to advance the global development of our product candidates. |
Cancer Immunotherapies and IFx Technology
The Cancer-Immunity Cycle
For an anti-cancer immune response to lead to effective killing of cancer cells a series of stepwise events must be initiated and allowed to proceed and expand iteratively. These steps, which are illustrated in the graphic below, are referred to as the “cancer-immunity cycle”. The human immune system is comprised of the innate immune system and adaptive immune system. The innate immune response, through evolution, has developed to protect us from our surrounding environment. It is the defense system with which we are born and serves as the body’s first defense mechanism against pathogens like bacteria or viruses and alerts the immune system to those threats. It works together with its complementary arm, the adaptive immune system, to address threats in the body, including cancer.
In the first step of the cycle, foreign proteins called “neoantigens” are created by cancer-related genes and are released and captured by dendritic cells (“DCs”) for processing. In order for this step to lead to a tumor killing T cell response, it must be accompanied by signals that specify immunity, or otherwise tolerance to the tumor antigens will be induced. Such immunogenic signals might include proinflammatory cytokines and factors released by dying tumor cells. During the next step, DCs present the captured neoantigens on MHCI and MHCII molecules to T cells, resulting in the priming and activation of tumor cell killing or cytotoxic, T cell responses against these cancer-specific neoantigens, which are viewed as foreign. Finally, the activated cytotoxic T cells traffic to and infiltrate the tumor bed, specifically recognizing and binding to cancer cells through the interaction between its T cell receptor (“TCR”) and its cognate antigen bound to MHCI and kill their target cancer cell. Killing of the cancer cell releases additional tumor-associated neoantigens repeating the first step of the cancer- immunity cycle, to increase the breadth and depth of the response in subsequent revolutions of the cycle.
In cancer patients, the cancer-immunity cycle does not perform optimally. In order for an innate response to be activated against a tumor, the tumor must appear foreign to the immune system. Tumor neoantigens may not be detected due to low neoantigen load or mutational burden, DCs and T cells may treat antigens as self rather than foreign thereby creating T regulatory cell responses rather than cytotoxic responses, T cells may not properly home to tumors, may be inhibited from infiltrating the tumor, or, importantly, factors in the tumor microenvironment might suppress those effector T cells that are produced. The goal of cancer immunotherapy is to initiate and reinitiate a self-sustaining cycle of cancer immunity, enabling it to amplify and propagate.
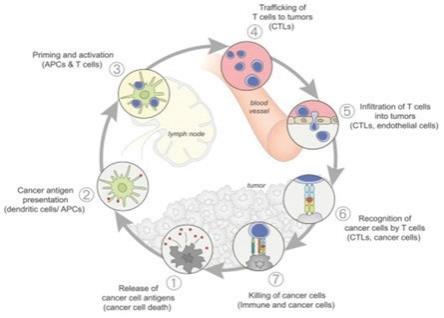
Source: Oncology Meets Immunology: The Cancer-Immunity Cycle, Immunity, Volume 39, July 2013
IFx Technology
The goal of cancer immunotherapies generally is to initiate an immune response to tumor neoantigens, which are the abnormal proteins that tumor-associated genetic mutations cause the cells to produce. There are a number of approaches that attempt to make a tumor look foreign to the immune system. The optimal cancer immunotherapy would make a patient’s entire tumor appear foreign and activate an innate immune response through the comprehensive and efficient packaging of tumor neoantigens which are presented to cytotoxic T cells, leading to their priming, activation, and proliferation of an immune attack against the tumor. TuHURA’s IFx Technology is designed to accomplish this goal.
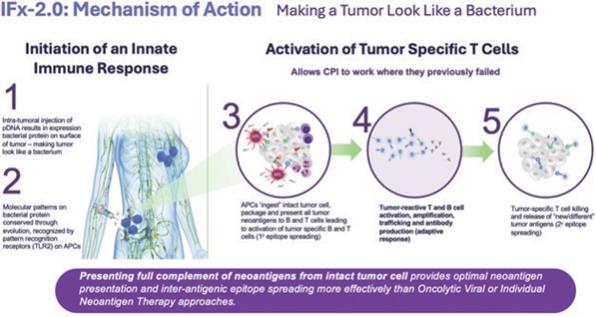
TuHURA’s IFx platform technology utilizes a proprietary plasmid DNA (“pDNA”) or messenger RNA (“mRNA”) which, when introduced into a tumor cell, results in the expression of a highly immunogenic gram positive bacterial protein (Emm55) from a rare variant of Streptococcus pyogenes on the surface of the tumor cell. This is graphically demonstrated above. By mimicking a bacterium, TuHURA’s technology makes a tumor cell look like bacteria. By making a tumor look like a bacterium, the molecular pattern of the bacterial protein is recognized by specific receptors on immune cells called pattern recognition receptors, also referred to as toll-like receptors or TLRs. These receptors are pre-programmed over evolution to recognize specific molecular patterns or motifs on pathogens like bacteria and activate and harness the power of the body’s innate immune response.
IFx is designed to harness the body’s natural innate immune response making the patients entire tumor appear foreign. This causes antigen presenting cells like DCs to phagocytize (which is the process of “eating” and “digesting”) the tumor cell, thinking they are bacteria. DCs present the captured neoantigens on MHCI and MHCII molecules to T cells, resulting in the priming and activation of cytotoxic T cell responses against these cancer-specific neoantigens, which are viewed as foreign. This is referred to as “primary epitope spreading.” Epitopes are the region/part of tumor antigens that are recognized by the immune system, specifically by antibodies, B cells and T cells. In doing so the first step of the cancer-immunity cycle is activated and restored.
Plasmid DNA, or plasmids, are small, circular, double-stranded DNA molecules that are separate from a cell’s chromosomal DNA and can replicate independently. Plasmids are most commonly found in bacteria, but can also be found in archaea and eukaryotic organisms. They can range in length from about 1,000 to hundreds of thousands of DNA base pairs. Plasmids often carry genes that can benefit the survival of an organism, such as antibiotic resistance. When a bacterium divides, all of the plasmids in the cell are copied, so each daughter cell receives a copy of each plasmid. Plasmids can also be transmitted horizontally to other bacteria in some cases. Scientists have taken advantage of plasmids to use them as tools to clone, transfer, and manipulate genes.
Other Types of Cancer Immunotherapies
To date, most cancer immunotherapies, such as those described below, have utilized a number of different approaches to initiate an innate immune response to generate tumor specific activated T cells.
Oncolytic Virus Vaccines. Oncolytic virus vaccines are designed to preferentially induce viral replication- dependent oncolysis (viral induced killing) in tumors in an effort to stimulate antitumor immune responses. Intratumoral injection is thought to trigger both local and systemic immunological responses leading to tumor cell lysis, the release of tumor-associated antigens into the tumor microenvironment where they need to be recognized by antigen presenting cells leading to subsequent activation of innate and adaptive immune systems to induce tumor antigen-specific effector T-cell antitumor immunity.
Tumor-associated antigen vaccines. Another approach is to utilize Tumor-Associated Antigens (“TAAs”), some of which may also be similar to self-antigens, although preferentially overexpressed on tumor cells. However, these TAAs may also be displayed by normal healthy cells or cancer testis antigens that are only expressed by tumor cells and adult reproductive tissues. T and B cells with high affinity toward these TAAs also target self-antigens leading to the removal of these T and B cells from the immune repertoire by central and peripheral tolerance. Thus, a potent vaccine must break tolerance for them to work. To date, this approach has had limited success.
Individual Neoantigen Therapy. Tumor-Specific Antigens (“TSAs”) differ from tumor-associated antigens since they are not shared with similar self-antigens. They are typically de novo epitopes expressed by cancer- causing viruses (or oncoviruses) or private neoantigens encoded by somatic mutations. TSAs are truly tumor specific with no central tolerance. Deciding which TSAs to select and how to configure such multivalent vaccines is itself a daunting challenge. It may be insufficient to rely entirely on sequencing the expressed tumor genome looking for point mutations, translocation fusions, or CT antigens. Not only might this vary from patient to patient or even from cell to cell within a single patient’s tumor, expression at the messenger RNA or protein
level does not assure that predicted antigenic peptides will be generated and expressed as peptide-MHCI complexes, especially in the face of the allelic complexity in the MHC. Several groups are actively approaching this problem by using a combination of informatics and mass spectroscopy of peptides eluted from MHCI molecules. Early clinical trials used as neo-adjuvant therapy in combination with checkpoint inhibitors among patients with potentially surgically curable disease at risk for relapse has yielded encouraging results, although how best to deliver them to patients remains a critical unknown.
Potential Advantages of IFx Innate Immune Agonist Technology
IFx’s approach is designed to naturally harness the power of the innate immune response leveraging Pathogen Associated Molecular Patterns (PAMP) or motifs present on pathogens, like bacteria and conserved through evolution. These patterns are recognized by pattern recognition receptors on antigen presenting and other immune cells of our innate immune system. By expressing a bacterial protein on the surface of a tumor cell the intact tumor cell is digested and the full complement of foreign tumor neoantigens are packaged and presented to newly produced T and B cells producing activated tumor specific T cells, the primary target allowing checkpoint inhibitors to work where they previously failed,
TuHURA believes that its IFx technology avoids problems associated with trying to predict which tumor- specific antigens are important and avoids the challenges associated with selection, analysis, production and delivery that accompanies individual neoantigen therapy approaches. Unlike oncolytic viral therapies which lyse the tumor cell disseminating tumor neoantigens throughout the tissue surrounding the tumor relying on antigen presenting cells in the vicinity to recognize, digest and present neoantigens to naïve T and B cells, IFx technology presents the full complement of tumor neoantigens from the intact tumor cell providing more optimal neoantigen presentation and inter-antigenic epitope spreading more effectively than oncolytic viral therapy or individual neoantigen therapy approaches.
Importantly, IFx is not an oncolytic viral technology. Oncolytic viral technologies which work by “exploding” the tumor cell resulting in the random dissemination of tumor neoantigens into the tumor microenvironment where immune cells can potentially see and digest them. In contrast, IFx presents the full complement of tumor neoantigens packaged inside the intact tumor cell providing much more optimal neoantigen presentation and more efficient inter-antigenic epitope spreading.
Clinical Rationale for TBS-2025
TBS-2025 (f/k/a KVA-12123), a VISTA inhibiting antibody, was initially investigated by Kineta in a large Phase 1 trial either as monotherapy (n=24) or in combination with pembrolizumab (n=15) among patients with advanced, therapy refractory cancers, including, breast, lung, colorectal and ovarian cancer. The drug demonstrated a favorable safety profile at the highest dose level of 1,000mg administered every two weeks. No significant anti-tumor activity was observed among the 39 patients treated in the trial.
VISTA is a novel checkpoint expressed on quiescent (resting) T cells and highly expressed on myeloid cells. While VISTA is expressed on a wide variety of solid tumor cancers, its role in resistance or failure of cancer- immunotherapy is not well established. Emerging scientific evidence demonstrates that mutNPM1 and mutDNM3TA, two of the most common mutations in AML and other myeloid (blood related) malignancies, drive the expression of VISTA on leukemic blasts in AML and are reported to be the primary mechanism by which AML has a poor response to and high relapse rate following current therapies. VISTA expression is linked to high relapse rate in AML due to its ability to allow leukemic blasts to evade immune recognition and attack by the patient’s immune system. When VSIR, the gene that encodes for VISTA is silenced, an immune response is observed and survival is enhanced in murine models of mutNPM1 AML
Recently, several new drugs called menin inhibitors have received accelerated approval in patients with relapsed and refractory mutNPM1. Menin is the “carrier” protein that exerts the proliferative effect on leukemic blasts. While the response rates of 25% to 30% that are seen following therapy with menin inhibitors are encouraging, they are short of short duration followed by leukemia recurrence and subsequent short survival. We believe adding TBS-2025 in treatment of patients with mutNPM1 r/r AML who are receiving a menin inhibitor may improve both response rate and duration of response by allowing immune recognition and attack against leukemic cells. We plan on investigating menin +/- TBS-2025 in mutNPM1 in r/r AML in a proof of concept study among 30 patients. If positive, this application of TBS-2025 would address an unmet medical need, and we believe it may qualify for development under the FDA’s accelerated approval pathway. We intend to start this Phase 2 randomized trial late in the fourth quarter of 2025.
DOR Technology and Bi-functional, bi-specific APCs and ADCs: Inhibiting MDSC immune suppressing functions
MDSCs
MDSCs are among the most common cells present in the tumor microenvironment, which is the tissue surrounding the tumor, where they are a major regulator of suppression of the immune system. MDSCs are normally produced during pregnancy where they migrate to and populate the placenta, creating an immunologic sanctuary for the fetus. Since half of the genetic make-up of the fetus comes from the father, this is necessary to prevent the mother’s immune system from attacking the fetus. They are also produced in settings of chronic inflammation or autoimmune disease as a mechanism to decrease inflammation or autoimmunity. Under normal conditions MDSCs represent less than 2% of circulating peripheral blood mononuclear cells (PBMCs) and lack potent immune suppressing characteristics.
In cancer, MDSCs are hijacked by tumors, being recruited to and creating an immunosuppressive environment in the tissues in which the tumor lives. Multiple effector molecules and signaling pathways are used by MDSCs to regulate immune suppression. One main mechanism involves depletion of necessary amino acids like arginine through production of arginase (“Arg-1”), or “destruction” of inflammatory cytokines via production of inducible nitric oxide (“iNOS”), in addition to anti-inflammatory prostaglandins (“COX2”), immune suppressing cytokines like transforming growth factor beta (“TGF-®”) or Interleukin 10 (“IL-10”) and recruitment and induction of immune inhibitory cells such as regulatory T cells (T regs) . In addition to these mechanisms to affect T cell functionality, they also exert a direct effect on T cells, thereby inhibiting their proliferation. Accumulating evidence demonstrates that the enrichment and activation of MDSCs correlates with tumor progression, metastasis and recurrence. In addition, MDSCs circulating in the blood of patients with cancer is highly correlated to poor clinical outcome.
We believe that inhibiting and reprograming MDSC function represents a promising novel approach to overcome MDSC-induced tumor microenvironment immunosuppression and acquired resistance to cancer immunotherapies. Various companies are focusing on several strategies, including blocking MDSC recruitment to the microenvironment or inhibiting their production in the bone marrow. Another potential strategy is inhibiting MDSC-mediated immunosuppression by developing inhibitors to individual MDSC-related immune suppressing compounds such as IDO, iNOS or COX2 inhibitors.
TuHURA’s Delta Opioid Receptor (DOR) inhibitors: bi-specific, bi-functional antibody peptide or drug conjugates (APC, ADCs)
The Delta Opioid Receptor, or DOR, is the first cloned G protein-coupled receptor. Many recent studies on Delta Opioid Receptor functions have determined that the Delta Opioid Receptor is involved in the regulation of malignant transformation and tumor progression in multiple cancers.
While Delta Opioid Receptor overexpression and its role in tumor biology has been studied, we believe that TuHURA, along with scientists at Moffitt Cancer Center, are the first to describe the high differential expression of the Delta Opioid Receptor on tumor-associated MDSCs compared to bone marrow (BM) or spleen derived MDSCs either in tumor free or tumor bearing models. (See figures below; source: TuHURA research files)

MDSC: MDSC isolated from BM, spleen and tumor. * p α 0.05, ** p α 0.01
As a previously unrecognized target to reprogram tumor associated MDSCs immunosuppressive functions on the tumor microenvironment, developing non-immunogenic peptidomimetic antagonists or small molecule antagonists with high specificity and avidity for the Delta Opioid Receptor represents a novel approach to reprograming MDSC functionality to overcome acquired resistance to checkpoint inhibitors and other cancer immunotherapies.
Inhibition of the Delta Opioid Receptor on tumor-associated MDSCs is designed to block MDSC production of multiple immunosuppressing factors through a single point of intervention. TuHURA’s bi-specific, bi- functional APCs consists, among other diverse candidates, a patented peptidomimetic Delta Opioid Receptor specific inhibitor conjugated to a checkpoint inhibitor like anti-PD-1 antibody. Moffitt Cancer Center scientists demonstrated that, in Delta Opioid Receptor-expressing PD-1 resistant murine lung cancer models, treatment with its APC accumulated in the tumor microenvironment and resulted in a significant and dramatic improvement in survival when compared to treatment with two times higher dose of the anti-PD-1 antibody alone. The Company has established multiple functional assay screens to investigate the effects of both novel peptidomimetic or small molecule Delta Opioid Receptor specific inhibitors of tumor-associated MDSC functionality to guide its selection of both APCs and ADCs for further invitro and in vivo characterization and development. The Company anticipates utilizing TBS-2025, its VISTA inhibiting antibody, as the first APC or ADC to enter preclinical development.
The Company believes that its tumor associated MDSC-targeting APCs and ADCs have a number of potential benefits over current approaches to overcoming acquired resistance to cancer immunotherapies, including the following:
| • | Inhibiting tumor associated MDSC production of multiple immune suppressing factors. The Delta Opioid Receptor on tumor-associated MDSCs functions like a “master switch” controlling the regulation of multiple immune suppressing factors such as, iNOS, Arg-1, IDO, TGF-b, S100A9 and COX2. Inhibiting the receptor results in “shutting off” production of these and other immune suppressing factors as compared to the industry focus of developing inhibitors targeting a single factor. |
| • | Blocking tumor-associated MDSC recruitment to the microenvironment. To exhibit their immunosuppressive phenotype, MDSCs have to be recruited to the tumor site, transitioning to tumor- associated MDSCs which display maximum immunosuppressive properties. This process is mediated mainly by chemokines secreted in the tumor microenvironment and chemokine receptors expressed on MDSCs. There are a number of strategies to prevent the recruitment of MDSCs to the microenvironment through the development of inhibitors of chemokines such as CCL2/CCR2 blockade. However, brain, heart, kidney, liver, lung, ovary, pancreas, spinal cord, spleen, and thymus also express CCR2, introducing the potential for off-target side effects with this approach. Inhibiting the Delta Opioid Receptor prevents the proliferation and production of tumor-associated MDSC- monocyte subpopulations (M-MDSC), promoting repolarizing M2 to M1 phenotype decreasing Th-2 cytokines while increasing Th-1 (g-IFN, IL-2) cytokines. Thus, changing the immunosuppressive phenotype of the tumor microenvironment to an immunogenic phenotype more favorable to cancer immunotherapies. |
| • | Immune modulation of tumor microenvironment/potentiating the effects of checkpoint inhibitors To date the prior and future development of ADCs, ADC-checkpoint inhibitors, ADC-degraders or bi- specific ADCs all have one thing in common: they target tumor-associated receptors with a tumor receptor-specific antibody which carries with it either a payload toxin or other tumor cell cycle disruptors or checkpoint inhibitor. To our knowledge, we are the only company developing APCs or ADCs targeting MDSCs where our APCs and ADCs are designed to be bi-specific/ bi-functional, i.e., affecting two targets and having two functions: inhibiting tumor associated MDSC-related immune suppression and thereby making tumor susceptible to attack, while localizing a checkpoint inhibitor, like TBS-2025, where the tumor resides. These two functions are intended to work together with the goal of overcoming acquired resistance, preventing T cell exhaustion and allowing checkpoint inhibitors and cellular therapies to be safer and more effective. |
TuHURA’s IFx Clinical Development Program
For purposes of the below descriptions of TuHURA’s Phase 1 and 1b clinical trials, the response rates for IFx-2.0 are determined under best clinical practice by the principal investigators, evaluating and confirming clinical progression prior to or during therapy utilizing conventional and appropriate radiographic or metabolic (Positron Emission Tomography – PET) methodologies. Response determination utilizes conventional terminologies under standardized response evaluation criteria. A “complete response”, or CR, is deemed to be disappearance of all lesions. A “partial response”, or PR, is at least a 30% decrease in the sum of the size of the target lesions. “Progressive disease”, or PD, is at least a 20% increase in the sum of the longest diameter or the appearance of new lesions. “Stable disease”, or SD, means that the patient has neither sufficient shrinkage in the lesions to qualify for PR nor sufficient increase to qualify for PD. The term “objective response rate”, or ORR, is defined as the proportion of patients who have a partial or complete response to therapy. Furthermore, the term “pCR” refers to a pathological complete response, which is the absence of signs of cancer in tissue samples removed during surgery or biopsy after treatment. “Progression-free survival”, or PFS, means the length of time after the treatment that a patient lives without disease progression.
Accelerated Approval Phase 3 Trial for IFx-2.0
TuHURA has entered into a Special Protocol Assessment agreement with the FDA for a single Phase 3 randomized placebo and injection controlled trial for IFx-2.0, its lead innate immune agonist, as adjunctive therapy to pembrolizumab (Keytruda®) in the first line treatment of patients with advanced or metastatic Merkel cell carcinoma, who are checkpoint inhibitor-naïve utilizing the FDA’s accelerated approval pathway. The Company has worked the deputy director of the FDA’s Oncology Center of Excellence (OCE) on a unique trial design. Consistent with the FDA’s Project Front Runner initiative, the FDA recommended the Company consider investigating IFx-2.0 in the front line treatment setting rather than in patients who are progressing on checkpoint inhibitor therapy, the latter of which was the conduct in the phase 1b trial. In doing so, data from a primary endpoint of objective response rate, or ORR, that is of sufficient magnitude and duration and with a favorable risk/benefit profile could be sufficient to support accelerated approval. ORR is considered to be a surrogate likely to predict clinical benefit, OCE requested that the Company also consider incorporating a key secondary endpoint that is not a surrogate for but an endpoint recognized to be of true clinical benefit such that results from a key secondary endpoint of progression-free survival, or PFS, that is adequately powered with statistical assumptions in the statistical analysis plan provided to the FDA, if achieved without a detrimental effect on overall survival, or OS, could be adequate to support conversion to regular approval satisfying the requirement for a confirmatory trial.
We anticipate that enrollment for the Phase 3 will take approximately 14 – 18 months, with topline data potentially being available 6 to 7 months following the last patient enrolled. If successful, this Phase 3 trial would form the basis of a Biologics License Application, or BLA. A Special Protocol Assessment agreement is a binding written agreement between the U.S. Food and Drug Administration (FDA) and a trial sponsor that indicates the FDA has agreed to the study’s design, charters, and statistical analysis plan and if the study endpoints are met within the context of the SPA Agreement such results would be adequate to support accelerated and regular approval. A Special Protocol Assessment agreement does not increase the likelihood of marketing approval for the product and may not lead to a faster or less costly development, review, or approval process. The study population, dose, schedule, and study design for the trial are based on the response rates observed in the Company’s Phase 1b trial in checkpoint inhibitor naïve patients with advanced Merkel cell carcinoma who exhibited primary resistance to anti PD(L)-1 checkpoint inhibitors such as Keytruda® The clinical study design for the Phase 3 registration trial is presented below. Based on correspondence with the FDA, patients with advanced Merkel cell carcinoma represent a patient population with an unmet medical need. TuHURA’s study, is designed to determine if IFx-2.0 can increase the objective response rate when used as adjunctive therapy to Keytruda in first line treatment of checkpoint inhibitor naïve patients with advanced Merkel cell carcinoma when compared to Keytruda alone.

Note: “FPI” means first patient in, “LPI” means last patient in, and “TLR” means top-line results. Progression Free Survival, or PFS, is defined as the time from randomization until first evidence of disease progression or death, and Overall Survival, or OS, is defined as the time between randomization to death.
Phase 1b Trial in Metastatic Merkel Cell Carcinoma and Cutaneous Squamous Cell Carcinoma
TuHURA has completed enrollment in a multicenter Phase 1b dose and schedule finding trial for TuHURA’s IFx-Hu2.0 innate immune agonist candidate in patients with advanced Merkel cell carcinoma or cutaneous Squamous cell carcinoma (cSCC). This study follows a two-stage design with a primary goal to assess the safety and feasibility of repeated dosing schemas of IFx-2.0. In the first stage (exposure escalation), a 3+3 trial design was utilized to assess safety of repeated weekly intratumoral injections using a fixed dose of IFx-2.0 weekly for 1, 2 or 3 weeks (for cohorts 1, 2 or 3 respectively). Following safety evaluation the protocol was amended to include an expansion stage to increase the total study sample size to 20. A total of 23 patients were enrolled. As of June 2024, follow-up data was available on all evaluable patients.
The primary objective of the trial was to determine the safety, tolerability, and optimal dose and schedule of IFx-2.0 when administered intratumoral in up to three lesions injected across three different administration schedules. Safety was evaluated for up to 28 days following IFx-2.0 administration. Secondary objectives include tumor shrinkage (injected and non-injected lesions) and correlative immune response analysis (transcriptomic, proteomic, humoral and cellular), pre-and post-IFx-2.0 administration to guide the choice of dose and schedule for the Company’s Phase 3 registration directed trial.
Twenty-three (23) patients were enrolled: Merkel cell carcinoma (13), cSCC (10). Among the thirteen (13) patients with Merkel cell carcinoma, twelve (12) completed treatment and the protocol directed 28 day safety evaluation follow up period; One (1) patient experienced a serious adverse event, or SAE, deemed possibly related to study drug. This patient experienced a Grade 3, or G3, adverse event, which is defined as an adverse event that is a severe or medically significant event that is not immediately life threatening, which in the case of this patient was a G3 autoimmune hepatitis that resolved with steroid treatment, and such patent has been recently treated with checkpoint inhibitors prior to study enrollment. Among the ten (10) patients with cSCC one (1) patient experienced an SAE unrelated to study drug and did not complete treatment nor the 28 day safety evaluation follow up period. All patients had received prior anti-PD(L)1 based treatment with disease progression being the reason for CPI discontinuation in all patients but one. Intra-tumoral (IT) IFx-2.0 was well tolerated at all dose schedules evaluated. As to efficacy, in the 21 patients that completed the study, best overall disease response to trial therapy was PR in 1 patient (including both injected and non-injected tumor sites), SD in 4, and PD in 16. The response assessment limited to the injected site(s) only was PR in 2 patients, SD in 8, and PD in 9. Two additional patients were not evaluable at the injected site(s) due to clinically challenging to measure dermal lesions that were not radiographically measurable. The study achieved the primary safety endpoint of the study demonstrating no grade 3 or greater toxicity in any of the 3 dose levels examined, and as a result, a recommended phase 2 dose was determined. The study also achieved its secondary endpoint of efficacy analysis demonstrating a disease control rate of 48% among injected lesions within the first 28 days post injection, and, as described below, a post-protocol efficacy analysis demonstrated an overall objective response rate of 64% (7 of 11 patients with Merkel cell carcinoma) after rechallenge with immune checkpoint inhibitors.
After protocol specified IT therapy, eleven (11) Merkel cell carcinoma patients and six (6) cSCC pts were treated with anti-PD(L)1 based therapy as the immediate post-protocol treatment. Five (5) of nine (9) (56%) evaluable Merkel cell carcinoma patients and one (1) of six (6) (17%) cSCC patients experienced an objective response to this ICI rechallenge, with duration of response ongoing in four (4) patients (6+, 19+, 21+, 23+ months) and the two other responses lasting 23 and 33 months. The two (2) remaining Merkel cell carcinoma patients were not evaluable for response from IO rechallenge due to radiation administered to the only measurable disease site(s), but both remain progression free at 11+ and 13+ months with previously progressive disease.
Of the twelve (12) patients with advanced Merkel cell carcinoma who completed treatment and protocol- directed 28-day safety evaluation follow-up period, seven patients exhibited primary resistance to first line treatment with a checkpoint inhibitor who did not receive subsequent therapies prior to receiving IFx-2.0. Five of seven patients received single agent anti-PD(L)-1 as initial therapy while two of seven patients received multiple CPIs as initial therapy including anti-PD-1, followed by anti-PD-1/anti-CTLA-4 therapy. All seven patients exhibited primary resistance to checkpoint inhibitor therapy progressing on average 3.3 months while receiving CPI therapy. These seven patients are graphically presented below:
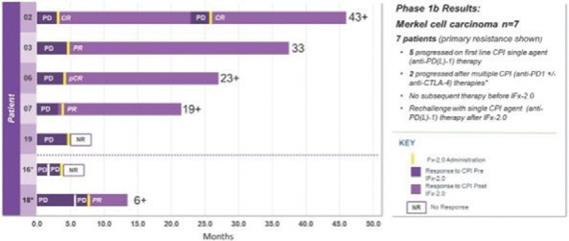
This data demonstrating the potential for IFx-2.0 to overcome primary resistance to anti-PD(L)-1 therapy and formed the clinical rationale for examining IFx-2.0 as adjunctive therapy with Keytruda® (anti-PD-1) in first line therapy among checkpoint inhibitor naïve patients with advanced or metastatic Merkel cell carcinoma. Unlike the phase 1b where IFx-2.0 was administered after patients progressing on anti-PD(L)-1 therapy, we believe IFx-2.0 could potentially provide a higher response rate to Keytruda® when administered prior to patients progressing failing Keytruda®.
The remaining seven (7) patients received multiple checkpoint inhibitor therapy including anti- CTLA-4/anti-PD-1 therapy and/or investigational agent(s) and or chemotherapy as 2nd or 3rd line therapy prior to treatment with IFx-2.0. This patient population is not representative of patients to be enrolled in the Phase 3 trial. Importantly, IFx-2.0 is not an intratumoral therapy like oncolytic viral therapies whose anti-tumor activity is limited to accessible, injected lesions in limited stages of cancer. In contrast, IFx-2.0’s mechanism of action is to prime and activate an innate immune response in injected lesions leading to a systemic anti-tumor response. The Company chose to examine IFx-2.0 in cutaneous malignancies because human skin has a high density of DCs which are very efficient in presenting foreign antigens to immune cells. Local injection of IFx-2.0 into cutaneous lesion(s) has resulted in immune cell infiltration, and in the context of MHCI and MHCII, tumor neoepitope presentation to naïve B and T cells followed by activation of tumor specific B and T cells. The immune response has not been localized to just injected lesions but rather systemic as demonstrated by production of Emm55 (pDNA encoded bacterial protein expressed on the surface of the tumor cell) and tumor specific IgM and IgG antibodies in the plasma of patients post IFx-2.0 administration.
Patients Merkel cell carcinoma-03 and Merkel cell carcinoma-05 below demonstrate the abscopal effect of adjunctive IFx-2.0 therapy, These patients exhibited primary resistance to checkpoint inhibitor therapy, and subsequently achieved durable anti-tumor responses following IFx-2.0 and rechallenge with checkpoint inhibitor therapy.
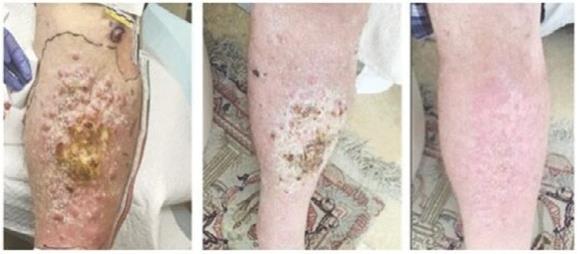
Case study (MCC-005)
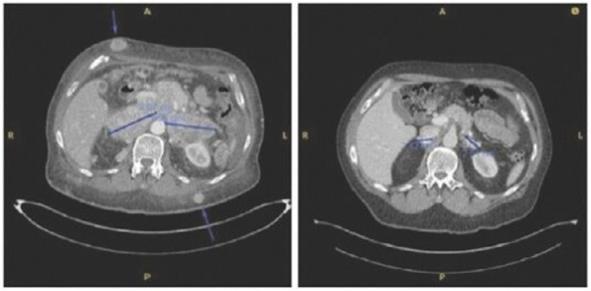
Patient was treated for multifocal in-transit recurrence of Merkel cell carcinoma in left leg with avelumab x 6 doses (12 weeks) with continued rapid clinical progression as well as development of liver metastatic disease on this therapy. Subsequently the patient was enrolled on IFx-2.0 protocol and received 3 weekly injections of IFx-2.0 without complication but continued clinical progression (additional in-transit sites). Disease status at time of last injection shown on the left. Following completion of IFx-2.0 protocol therapy, subject was rechallenged with pembrolizumab, a checkpoint inhibitor, and experienced an obvious clinical response initially apparent approximately 3-4 weeks into therapy. Clinical response at 3 months (middle photo below) and 6 months (right photo below) are shown in the photos below. Concordant (near-complete) radiographic response of liver metastases has also been observed and response has been maintained to date (19 months)
Case study (MCC-002)
Subject was treated with adjuvant pembrolizumab for stage II Merkel cell carcinoma on the STAMP trial but developed (nodal) progression after receiving 6 doses. Subject underwent salvage surgery/XRT but developed widespread metastatic disease ~3 months later (nodal, dermal, and intramuscular sites of disease). Subject was then enrolled on IFx-2.0 protocol and received 2 weekly injections to 3 nodal/dermal metastatic sites but experienced continued rapid progression (both injected and non-injected sites) including bulky diffuse adenopathy and numerous widespread subcutaneous/dermal nodules. Representative imaging from the time of completion of protocol therapy is shown on left in photo below including several subcutaneous sites (as noted by the arrows) and bulky retroperitoneal (“RP”) conglomerate lymph node (“LN”) metastases. Post-protocol, subject was started on checkpoint inhibitor rechallenge with avelumab and experienced deep partial response that has been maintained to date (33 months). Representative images from post-checkpoint rechallenge restaging shown below on right (complete remission of subcutaneous nodules, partial response in retroperitoneal sites).
IFx-2.0 Phase 1b/2a Study of IFx-Hu2.0 as an Adjunctive Therapy to Keytruda® (pembrolizumab) in First Line Treatment for Metastatic Merkel Cell Carcinoma of Unknown Primary Origin (MCCUP)
In May 2025, we initiated a Phase 1b/2a trial designed to evaluate the safety and feasibility of IFx-Hu2.0 in combination with Keytruda® when administered via Interventional Radiology (IR) in patients with deep- seated tumors without associated cutaneous tumors. Unlike our Phase 3 study, these are patients without skin lesions who present with metastatic deep-seated tumors in the liver, lungs or retropertitoneum (abdomen). Up to 30% of patients with MCC present without primary lesions in the skin, so this trial will not only provide safety, feasibility, and efficacy data, but may also expand the potential number of addressable patients who may benefit from IFx-Hu2.0,
If feasibility and safety is demonstrated for IFx-Hu2.0 and Keytruda® when radiologically administered to deep-seated tumors, we plan to extend enrollment to a variety of non-MCC cancers that are known not to respond or respond poorly to CPIs. This is termed a “Basket Trial.” Since the underlying biology of why tumors don’t respond to CPIs is for the most part the same, then we believe that the mechanism of how IFx-Hu2.0 overcomes that resistance to CPIs should be independent of the type of cancer treated. We have previously demonstrated that IFx-Hu2.0 can overcome CPI resistance in melanoma, squamous cell, and Merkel cell carcinoma, three unrelated types of skin cancers. If successful, this trial could expand the potential benefit of IFx-Hu2.0 to a wide variety of cancers.
Phase 1 Trial in Advanced, (Stage IIIC-IV) Cutaneous Melanoma
TuHURA has also conducted a Phase 1 trial at the Moffitt Cancer Center in seven (7) patients with advanced (Stage IIIc/IV) cutaneous melanoma, six (6) of whom were eligible for evaluation post-IFx-2.0 therapy. The primary objective of the trial was to determine the safety and tolerability of IFx-2.0 when administered intratumorally with up to three lesions injected at a single time point. Safety was evaluated for 28 days following IFx-2.0 administration. Secondary objectives included tumor shrinkage, transcriptomic, proteomic, humoral, and cellular immune response pre and post IFx-2.0 administration. IFx-2.0 was well tolerated. Mild pain and swelling among injected lesions were most common reported side effect < Grade 2 in severity. Four (4) of the six (6) patients exhibited primary resistance to, and failed checkpoint inhibitor trials prior to IFx-2.0. Following IFx-2.0 administration three (3) of four (4) patients subsequently responded to rechallenge with checkpoint inhibitor(s). One patient achieved stable disease (“SD”) and 2 experienced a partial response (“PR”). As of the last follow up responses are ongoing at 1337, 608, 313 days. Two (2) patients (SD and PR) underwent surgical
resections following checkpoint inhibitor therapy. Immunologic profiling data (pre-and post-IFx-2.0) demonstrated a robust systemic immune response with (i) activation of tumor specific B cells with tumor specific IgM/IgG antibody production recognizing hundreds of previously unrecognized melanoma tumor neoepitopes and (ii) gene signature, consistent with innate response in injected lesions, a gene signature consistent with adaptive response in un-injected lesions as well as increased expression (up to 11 fold) of genes known to be predictive of response to checkpoint inhibitors following IFx-2.0 therapy but prior to checkpoint inhibitor rechallenge.
TuHURA’s TBS-2025 VISTA Inhibiting Antibody Clinical Development Program
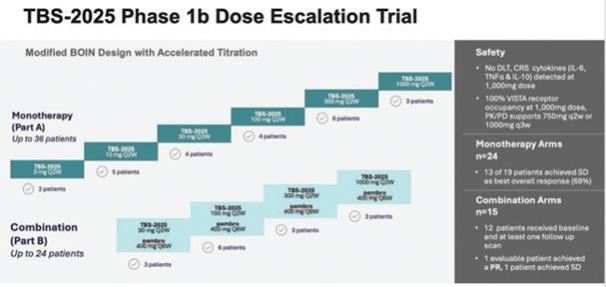
TBS-2025 (f/k/a KVA-12123), a VISTA inhibiting antibody, was initially investigated by Kineta in a large Phase 1 trial either as monotherapy (n=24) or in combination with pembrolizumab (n=15) among patients with advanced, therapy refractory cancers, including, breast, lung, colorectal and ovarian cancer. The Phase 1 was an open-label, multi-center, dose-escalation trial, utilizing an accelerated Bayesian Optimal Interval (BOIN) dosing design designed to evaluate the safety, tolerability, pharmacokinetics (“PK”), immunogenicity, and tumor response of TBS-2025. TBS-2025 demonstrated a favorable safety profile at the highest dose level of 1,000 mg administered every two weeks. In this trial among patients with treatment-refractory solid tumors, no significant anti-tumor activity was observed among the 39 patients treated in the trial.
An overview of the study results is shown below:
Clinical collaboration with Merck
Kineta previously entered into a clinical trial collaboration and supply agreement with Merck (known as MSD outside the U.S. and Canada) that we have assumed as a part of the Kineta acquisition. Under this collaboration, we are evaluating the safety, tolerability, PK. and anti-tumor activity of TBS-2025 alone and in combination with KEYTRUDA® (pembrolizumab), Merck’s anti-PD-1 therapy, in patients with advanced solid tumors.
Pharmacokinetics (PK) and Receptor Occupancy (RO)
Pharmacokinetics, or PK, is the study of how the body interacts with TBS-2025 for the entire duration of exposure after administration. TBS-2025 exhibited a greater than dose-proportional pharmacokinetic profile in drug exposure across all doses, consistent with target-mediated drug disposition at lower doses and target saturation at higher doses.
To guide the recommended Phase 2 dose decision, Kineta developed a proprietary assay to evaluate VISTA receptor occupancy (“RO”) on immune cells from patients treated with TBS-2025. This is an important metric for evaluating how well TBS-2025 is blocking the VISTA target. TBS-2025 achieved a greater than 90% VISTA RO at the 30 mg dose and a complete saturation of the target between two-dosing intervals was achieved at 1000 mg. Based on these data. the Company believes the Recommended Phase 2 Dose (RP2D) should be 750mg every two weeks.
Biomarkers
In drug development and clinical trials, biomarkers may be useful to identify patient populations for a study, monitor therapeutic response, and identify side effects. TBS-2025 demonstrated dose-proportional on-target biomarker immune responses involved in anti-tumor activity. TBS-2025 demonstrated significant efficacy- related, dose-dependent cytokine induction of CXCL10, IFNα, CCL2 (MCP1), CCL3 (MIP1α), CCL4 (MIP1ß) and CXCL8 (IL8), which are involved in immune cell activation and recruitment to the tumor microenvironment. Additionally, increases in anti-tumor immune cell subpopulations including nonclassical monocytes with an activated phenotype (increased of cell surface expression of HLA-DR and CD80), NK cells, CD4+ T cells and CD8+ T cells were observed during treatment.
TBS-2025 demonstrated induction of pro-inflammatory myeloid-derived cytokines/chemokines involved in immune cell activation and recruitment in the tumor microenvironment. Changes in these key biomarkers and immune cell populations are indicative of the anti-tumor effects of blocking VISTA that is consistent with data from preclinical models (NHP and KO mice). These data validate their use as potential biomarker of VISTA target engagement with TBS-2025
Phase 2 Randomized controlled trial of menin inhibitor +/- TBS-2025 in mutNPM1 r/r AML
VISTA is a novel checkpoint expressed on quiescent (resting) T cells and highly expressed on myeloid cells. While VISTA is expressed on a wide variety of solid tumor cancers, its role in resistance or failure of cancer- immunotherapy is not well established. Emerging scientific evidence demonstrates that mutNPM1 and mutDNM3TA, two of the most common mutations in AML and other myeloid (blood related) malignancies, drive the expression of VISTA on leukemic blasts in AML and are reported to be the primary mechanism by which AML has a poor response to and high relapse rate following current therapies. VISTA expression is linked to high relapse rate in AML due to its ability to allow leukemic blasts to evade immune recognition and attack by the patient’s immune system. When VSIR, the gene that encodes for VISTA is silenced, an immune response is observed, and survival is enhanced in murine models of mutNPM1 AML
Recently, several new drugs called menin inhibitors have received accelerated approval in patients with relapsed and refractory mutNPM1. Menin is the “carrier” protein that exerts the proliferative effect on leukemic blasts. While the response rates of 25% to 30% that are seen following therapy with menin inhibitors are encouraging, they are short of short duration followed by leukemia recurrence and subsequent short survival. We believe adding TBS-2025 in treatment of patients with mutNPM1 r/r AML who are receiving a menin inhibitor may improve both response rate and duration of response by allowing immune recognition and attack against leukemic cells. We plan on investigating menin +/- TBS-2025 in mutNPM1 in r/r AML in a proof of concept Phase 2 trial among 30 patients. If positive, this application of TBS-2025 would address an unmet medical need, and we believe it may qualify for development under the FDA’s accelerated approval pathway. We intend to start this Phase 2 randomized trial late in fourth in the fourth quarter 2025.
TuHURA’s Manufacturing Strategy
TuHURA maintains established relationships with contract development and manufacturing organizations (CDMOs) to manufacture and test IFx-Hu2.0 clinical trial material (“CTM”), including drug substance and drug products required for registration trials.
IFx-Hu2.0 is comprised of 1) the Plasmid DNA (pAc/emm55) in TE Buffer Drug Product (DP) with 10% Dextrose Injection. and 2) the Cationic Polymer DP with 10% Dextrose Injection. The Plasmid DNA (pAc/ emm55) in TE Buffer DP utilizes the Cationic Polymer DP as a transfectant agent excipient, and IFx-Hu2.0 is complexed at the site prior to patient administration. TuHURA has completed the FDA-required mixing studies demonstrating the mixing process consistently produces a product that meets a set of quality attributes. IFx- Hu2.0 preparation instructions are included in the pharmacy manual to ensure mixing at the site prior to administration results in reliably produced drug product with consistent material properties. In addition, the FDA-required potency and stability assays have been developed, qualified, and/or validated supporting product release and stability, which meets cGMP requirements for use in our Phase 3 registration trial.
TuHURA assumed from Kineta a manufacturing agreement with Samsung Biologics to provide manufacturing services, including CTM drug substance and drug product manufacturing and stability testing for TBS-2025. Samsung has no commercial rights to TBS-2025 or any other assets acquired from Kineta.
Intellectual Property
Intellectual property is of vital importance in TuHURA’s field and in biotechnology generally. The company seeks to protect and enhance proprietary technology, inventions, and improvements that are commercially important to the development of TuHURA’s—business by seeking, maintaining, and defending patent rights, whether developed internally or licensed from third parties. TuHURA also seeks to rely on regulatory protection afforded through inclusion in expedited development and review, data exclusivity, market exclusivity and patent term extensions where available. TuHURA has sought patent protection in the United States and internationally related to its IFx-Hu2.0 platform technology as well as its IFx-Hu3.0 technology, and TuHURA licenses from third parties the patents and patent applications relating to its tumor microenvironment modulators technology.
TuHURA expects to file additional patent applications in support of current and new clinical candidates, as well as new platform and core technologies. TuHURA’s commercial success will depend in part on obtaining and maintaining patent protection and trade secret protection of TuHURA’s current and future product candidates and the methods used to develop and manufacture them, as well as successfully defending any such patents against third-party challenges and operating without infringing on the proprietary rights of others. TuHURA’s ability to stop third parties from making, using, selling, offering to sell or importing its product candidates will depend on the extent to which TuHURA has rights under valid and enforceable patents or trade secrets that cover these activities.
The terms of individual patents depend upon the statutory term of the patents in the countries in which they are issued. In most countries in which TuHURA files, including the United States, the patent term is 20 years from the earliest filing of a non-provisional patent application. In the United States, a patent term may be lengthened by patent term adjustment (“PTA”), which compensates a patentee for administrative delays by the USPTO in examining and granting a patent. Conversely, a patent term may be shortened if a patent is terminally disclaimed over an earlier filed patent. In the United States, the term of a patent that covers an FDA-approved drug may also be eligible for extension, which permits patent term restoration to account for the patent term lost during the FDA regulatory review process. The Hatch-Waxman Act permits a patent term extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the subject drug candidate is under regulatory review. Patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval, only one patent applicable to an approved drug may be extended and only those claims covering the approved drug, a method for using it, or a method for manufacturing it may be extended. Similar provisions to extend the term of a patent that covers an approved drug are available in Europe and other foreign jurisdictions. In the future, if and when TuHURA’s products receive FDA approval, TuHURA expects to apply for patent term extensions on patents covering those products. TuHURA plans to seek patent term extensions to any issued patents TuHURA may obtain in any jurisdiction where such patent term extensions are available, however there is no guarantee that the applicable authorities, including the FDA in the United States, will agree with TuHURA’s assessment that such extensions should be granted, and if granted, the length of such extensions.
In some instances, TuHURA has submitted and expects to submit patent applications directly to the USPTO as provisional patent applications. Corresponding non-provisional patent applications must be filed not later than 12 months after the provisional application filing date. While TuHURA intends to timely file non-provisional patent applications relating to TuHURA’s provisional patent applications, TuHURA cannot predict whether any such patent applications will result in the issuance of patents that provide TuHURA with any competitive advantage.
TuHURA expects to file U.S. non-provisional applications and Patent Cooperation Treaty, or PCT, applications that claim the benefit of the priority date of earlier filed provisional applications, when applicable. The PCT system allows a single application to be filed within 12 months of the original priority date of the patent application and to designate all of the PCT member states in which national patent applications can later be pursued based on the international patent application filed under the PCT. A designated authority performs an initial search and issues a non-binding opinion as to the patentability of the subject matter. The opinion may be used to evaluate the chances of success of national phase applications in various jurisdictions, thereby informing the development of a global filing strategy.
Although a PCT application does not itself issue as a patent, it allows the applicant to conveniently file applications in any of the member states through national-phase applications. At the end of a period of 30-31 months from the earliest priority date of the patent application (varies by jurisdiction), individual applications can be filed in any of the PCT member states/regions. Use of the PCT system is more cost-effective than direct foreign filings and permits applicants greater flexibility with respect to budgeting and the selection of foreign jurisdictions.
For all patent applications, TuHURA determines claiming strategy on a case-by-case basis. Advice of counsel and TuHURA’s business model and needs are always considered. TuHURA seeks to file patents containing claims for protection of all useful applications of TuHURA’s proprietary technologies and any products, as well as all new applications and/or uses TuHURA discovers for existing technologies and products, assuming these are strategically valuable. TuHURA continuously reassess the number and type of patent applications, as well as the pending and issued patent claims to pursue maximum coverage and value for TuHURA’s processes, and compositions, given existing patent office rules and regulations. Further, claims may be modified during patent prosecution to meet TuHURA’s intellectual property and business needs.
TuHURA recognizes that the ability to obtain patent protection and the degree of such protection depends on a number of factors, including the extent of the prior art, the novelty and non-obviousness of the invention, and the ability to satisfy the enablement requirement of the patent laws. In addition, the coverage claimed in a patent application can be significantly reduced before the patent is issued, and its scope can be reinterpreted or further altered even after patent issuance. Consequently, TuHURA may not obtain or maintain adequate patent protection for any of TuHURA’s future product candidates or for TuHURA’s technology platform. TuHURA cannot predict whether the patent applications it is currently pursuing will issue as patents in any particular jurisdiction or whether the claims of any issued patents will provide sufficient proprietary protection from competitors. Any patents that TuHURA holds may be challenged, circumvented or invalidated by third parties.
The patent positions of biotechnology companies are generally uncertain and involve complex legal, scientific and factual questions. TuHURA’s commercial success will also depend in part on not infringing upon the proprietary rights of third parties. Third-party patents could require TuHURA to alter its development or commercial strategies, or TuHURA’s products or processes, obtain licenses or cease certain activities. TuHURA’s breach of any license agreements or its failure to obtain a license to proprietary rights required to develop or commercialize TuHURA’s future products may have a material adverse impact on the company.
If third parties prepare and file patent applications in the United States that also claim technology to which TuHURA has rights, TuHURA may have to participate in interference or derivation proceedings in the USPTO to determine priority of invention. For more information, see “Risk Factors — Risks Relating to TuHURA – Risks Relating to TuHURA’s Intellectual Property.”
When available to expand market exclusivity, TuHURA’s strategy is to obtain, or license additional intellectual property related to current or contemplated development platforms, core elements of technology and/ or clinical candidates.
Company-owned Intellectual Property
As of September 11, 2025, TuHURA had 33 issued patents over 13 jurisdictions, and 9 pending applications (2 U.S. utility patent applications and 7 foreign patent applications). Most of such patents and patent applications relate to TuHURA’s IFx technology platform. The following is a summary of TuHURA’s issued patents and pending patent applications as of September 11, 2025 by patent family.
| Patent Family |
Description |
Application/ |
Filing Date |
Issue Date/Status |
Earliest Expected Date |
Type of Parent | ||||||
| DNA Vector and Transformed Tumor Cell Vaccines |
Whole cell and DNA cancer vaccines | PCT/US2015/018688 (WO 2015/134577) | 03/04/2015 | Nationalized in CH, DE, DK, EP, FR, GB, HK, IE, NL, NO, SE, US |
3/4/2035 | Use Composition Composition | ||||||
| US 9,555,088 US 9,839,680 US 10,391,158 US 10,751,400 |
07/07/2016 01/30/2017 12/11/2017 08/26/2019 | Issued 01/31/2017 Issued 12/12/2017 Issued 08/27/2019 Issued 08/25/2020 | 3/4/2035 3/4/2035 3/4/2035 3/4/2035 |
Use Composition Composition Method | ||||||||
| Cancer Vaccine Comprising mRNA Encoding a M-Like-Protein |
Next generation cancer vaccine using mRNA encoding a bacterial antigen to prime anti-cancer immune responses | PCT/US2016/033235 (WO 2016/187407) | 05/19/2016 | Nationalized in AU, CA, CH, CN, DE, DK, EP, FR, GB, HK, IE, JP, NL, NO, SE, US |
5/19/2036 | Use Composition Composition/use | ||||||
| US 9,636,388 US 10,682,401 US 18/060,605 |
07/28/2016 05/01/2017 12/01/2022 | Issued 05/02/2017 Issued 06/16/2020 pending | 5/19/2036 5/19/2036 5/19/2036 |
Use Method Composition | ||||||||
| Modified mRNA for Multicell Transformation |
Next generation cancer vaccine using mRNA encoding a bacterial antigen to prime anti-cancer immune responses | PCT/US2021/031204 (WO 2021/226413) | 5/7/2021 | Nationalized in CN, JP, CA, IN, AU, EP, KR, HK | 5/7/2041 | |||||||
Intellectual Property Acquired in Kineta Merger
As of September 11, 2025, our patent portfolio acquired from Kineta as it pertains to TBS-2025 included fourteen (14) national phase applications in the KVA-001 patent family related to TBS-2025. The countries are as follows: U.S., Australia, Brazil, Canada, China, Europe (European Patent Office (“EPO”)), Hong Kong, Israel, India, Japan, Korea, Mexico, Russia, and Singapore. Its estimated expiration date without any patent term adjustment or extension is 20 years from filing, i.e., February 18, 2042.
The table below summarizes the high-level filing strategy of our existing patent portfolio for the TBS-2025 related assets acquired from Kineta:
| VISTA patents (TBS-2025 f/k/a KVA12123) | ||
| Patent Family |
KVA-001 | |
| Composition of matter |
Y | |
| Methods of Manufacturing |
Y | |
| Sequences/Structure |
Y | |
| Indications |
Y | |
| Specification on use (mono or combo) |
Y | |
| Binding characteristics |
Y | |
| Immune cell regulation |
Y | |
| Physiologic properties |
Y | |
| Discovery Candidates |
To be added on a rolling basis |
Kineta strives to protect the proprietary technologies that it believes are important to its business, including by seeking, maintaining and defending patent rights, whether developed internally or in conjunction with or in- licensed from third parties. Kineta also relies on trade secrets relating to its monoclonal antibodies, know-how, continuing technological innovation and in-licensing opportunities to develop, strengthen and maintain its proprietary position in the field of innate immunity and fully human antibodies.
As more fully described above, as of September 11, 2025, our patent portfolio related to TBS-2025 included 14 U.S. and foreign applications, which entered national phase in 2023.
Licensed Intellectual Property Rights Relating to DOR Technology
TuHURA licenses the intellectual property rights relating to its DOR technology platform under exclusive license agreements with H. Lee Moffitt Cancer Center and Research Institute (“Moffitt Cancer Center”) and the West Virginia University Research Corporation (“WVURC”). In particular, TuHURA is a party to a March 2019 Exclusive License Agreement with Moffitt Cancer Center under which, as amended, we license patent rights co- owned by Moffitt and University of South Florida relating to ADCs for immunotherapy and Delta receptor targeted agents for molecular imaging and immunotherapy of lung cancer. TuHURA is a party to a second Exclusive License Agreement entered into in April 2021 under which, as amended, we license Moffitt’s interest in certain patent rights relating to the applicability of TuHURA’s Delta receptor technology to the tumor microenvironment (these patent rights are co-owned by Moffitt and us). TuHURA is a party to a September 2022 Restated and Amended Exclusive License Agreement with WVURC pursuant to which TuHURA licenses from WVURC certain patent rights (including WVURC’s rights under one patent that is jointly owned by WVURC and the company) relating to Delta receptor targeted agents for molecular imaging and cancer immunotherapy. These license agreements were originally entered into with Moffitt and WVURC by TuHURA Biopharma which assigned its interest under the agreements to TuHURA as a part of the acquisition of certain TuHURA Biopharma assets in January 2023. The following are summaries of the material terms of these license agreements:
2019 License Agreement with Moffitt Cancer Center
In March 2019, TuHURA Biopharma, as predecessor in interest to the company, entered into an Exclusive License Agreement with Moffitt Cancer Center, which agreement was amended in September 2019, April 2021 and August 2022 (as amended, the “2019 Moffitt Agreement”), for the worldwide, exclusive license of patents for the development, commercialization and marketing of products derived from Moffitt’s rights to patents entitled “Conjugates for Immunotherapy” and “A Delta-Opioid Receptor Targeted Agent For Molecular Imaging And Immunotherapy Of Lung Cancer” (the “2019 Moffitt Licensed Patents”). The exclusive nature of the
granted licenses are subject to customary reservations by Moffitt for non-commercial research, development, and academic purposes. The licenses granted by Moffitt are sublicensable by TuHURA to affiliates and third parties, subject to certain requirements, including providing Moffitt with a copy of each executed sublicense agreement and ensuring that the sublicensee complies with the terms of the 2019 Moffitt Agreement.
Pursuant to the terms of the 2019 Moffitt Agreement, in partial consideration of Moffitt’s grant of the rights and licenses, TuHURA Biopharma paid to Moffitt one-time, non-refundable license issue fees of $100,000 and $30,000. Additionally, TuHURA Biopharma issued shares of its Common Stock to Moffitt as additional consideration, which were exchanged for 146,397 shares of the company’s Common Stock (after giving effect to the exchange ratio in the Kintara Merger) as a part of the TuHURA Biopharma asset acquisition. The company is obligated to pay Moffitt an annual license maintenance fee not in excess of $50,000 per year until annual minimum royalty payments commence following commercial sales of licensed products.
Also under the 2019 Moffit Agreement, TuHURA is required to make the following additional payments:
| • | Various milestone royalty payments based on specified development, approval, commercialization, and sales milestones, which payments range from $150,000 to $400,000 for milestones relating to the commencement of clinical trials up to $3.0 million to $5.0 million based on sales thresholds in excess of $1.0 billion in sales; |
| • | Running royalties based on net sales of licensed products with a royalty percentage in the middle-single digit and with escalating minimum annual royalties that do not exceed $0.5 million per year; and |
| • | Payment of all patent prosecution and maintenance costs and fees for the licensed patents. |
The term of the 2019 Moffitt Agreement will be until the later of (i) the date on which the last of the licensed patents expire, or (ii) twenty (20) years after the date of the 2019 Moffitt Agreement. TuHURA may unilaterally terminate the 2019 Moffitt Agreement at any time on six (6) months’ notice to Moffitt, provided that all payments due by TuHURA at that time have been made through the effective date of termination. Additionally, TuHURA may terminate the agreement with written notice to Moffitt in the event Moffitt commits a material breach and such breach is not cured within sixty (60) days following Moffitt’s receipt of such notice. Moffitt has the right to terminate, or convert all exclusive licenses to nonexclusive licenses in the event TuHURA: (x) fails to make payments due under the agreement within thirty (30) days following notice from Moffitt; (y) commits a material breach that is not cured, or capable of being cured, within sixty (60) days after receipt of notice from Moffitt; (z) or challenges the validity of any of the 2019 Moffitt Licensed Patents before a court or other administrative agency in any jurisdiction. Upon any termination prior to the expiration of the agreement for any reason, all licenses and rights granted pursuant to the agreement will automatically terminate. At the request of Moffitt, TuHURA is obligated to provide all materials, clinical results, regulatory submissions, registrations and any other related filings for the 2019 Moffitt Licensed Patents, and all data used to support the same, to Moffitt.
2021 License Agreement with Moffitt Cancer Center
In April 2021, TuHURA Biopharma, as predecessor in interest to the company, entered into an Exclusive License Agreement with Moffitt, which agreement was amended in August 2022 (collectively, the “2021 Moffitt Agreement”), for the worldwide, exclusive, license to Moffitt’s rights under a jointly-owned patent entitled “Delta Opioid Receptor Antagonist Reprogram Immunosuppressive Microenvironment to Boost Immunotherapy” (the “2021 Moffitt Licensed Patent”) for the development, commercialization and marketing of products from covered claims of the 2021 Moffitt Licensed Patent. The exclusive nature of the licenses granted are subject to customary reservations by Moffitt for non-commercial research, development, and academic purposes. The licenses granted by Moffitt are sublicensable by the company to affiliates and third parties, subject to certain requirements, including providing Moffitt with a copy of each executed sublicense agreement, and ensuring that the sublicensee comply with the terms of the 2021 Moffitt Agreement.
Pursuant to the terms of the 2021 Moffitt Agreement, in partial consideration of Moffitt’s grant of the rights and licenses, TuHURA Biopharma paid to Moffitt a one-time, non-refundable license issue fee of $12,500. Additionally, TuHURA Biopharma issued shares of its Common Stock to Moffitt as additional consideration, which were exchanged for 195,465 shares of the company’s Common Stock as a part of the TuHURA Biopharma asset acquisition. TuHURA is obligated to pay Moffitt an annual license maintenance fee not in excess of $25,000 per year until annual minimum royalty payments commence following commercial sales of licensed products.
TuHURA is also required to make the following additional payments:
| • | Various milestone royalty payments based on specified development, approval, commercialization, and sales milestones, which payments range from $37,500 to $100,000 for milestones relating to the commencement of clinical trials up to $750,000 to $1.25 million based on sales thresholds in excess of $1.0 billion in sales; and |
| • | Running royalties based on net sales of licensed products with a royalty percentage in the middle-single digit and with escalating minimum annual royalties that do not exceed $0.1 million per year; and |
| • | Payment of all patent prosecution and maintenance costs and fees for the licensed patents. |
The term of the 2021 Moffitt Agreement will be until the later of (i) the date on which the last of the patents expire, or (ii) twenty (20) years after the date of the 2021 Moffitt Agreement. TuHURA may unilaterally terminate the 2021 Moffitt Agreement at any time on six (6) months’ notice to Moffitt, provided that all payments due by TuHURA at that time have been made through the effective date of termination. Additionally, TuHURA may terminate the agreement with written notice to Moffitt in the event Moffitt commits a material breach and such breach is not cured within sixty (60) days following Moffitt’s receipt of such notice. Moffitt has the right to terminate, or convert all exclusive licenses to nonexclusive licenses in the event we: (x) fail to make payments due under the agreement within thirty (30) days following notice from Moffitt; (y) commit a material breach that is not cured, or capable of being cured, within sixty (60) days after receipt of notice from Moffitt; (z) or challenge the validity of any of the 2021 Moffitt Licensed Patent before a court or other administrative agency in any jurisdiction. Upon any termination prior to the expiration of the agreement for any reason, all licenses and rights granted pursuant to the agreement will automatically terminate. At the request of Moffitt, TuHURA is obligated to provide all materials, clinical results, regulatory submissions, registrations and any other related filings for the 2021 Moffitt Licensed Patent, and all data used to support the same, to Moffitt.
License Agreement with West Virginia University Research Corporation
In January 2023 but with an effective date of September 2022, TuHURA Biopharma, as predecessor in interest of the company, entered into a Restated and Amended Exclusive License Agreement with WVURC (the “WVU Agreement”), which terminated and replaced the prior agreement between WVURC and TuHURA Biopharma. The WVU Agreement provides for the exclusive commercialization rights relating to Delta receptor targeted agents for WVURC patent rights relating to molecular imaging and cancer immunotherapies (the “WVU Patents”). Under the WVU Agreement, among other rights, WVURC granted TuHURA a worldwide, exclusive right, with limited sublicense rights, to develop and commercialize the WVU Patents in accordance with the milestone schedule therein.
As partial consideration for the rights granted under the WVU Agreement, TuHURA Biopharma previously paid a non-refundable, upfront fee of $50,000. Under the terms of the WVU Agreement, TuHURA is required to pay WVURC a tiered running royalty in the low-to-mid single digit percentages based on levels of net sales of licensed products, including the net sales of sublicensees, with customary anti-stacking provisions. TuHURA is also required to pay annual fees of $30,000 or less and is required to fund all patent prosecution and maintenance costs and fees for the licensed patents.
The term of the WVU Agreement will expire on the later of: (i) the expiration of the date of the last to expire of the WVU Patents or (ii) twenty (20) years from the first commercial sale of a licensed product derived from the WVU Patents, unless earlier terminated pursuant to its terms. TuHURA may unilaterally terminate the WVU Agreement upon written notice to WVURC at any time on six (6) months’ notice to WVURC, provided that all payments due by TuHURA at that time have been made through the effective date of termination. Additionally, TuHURA may terminate the agreement with written notice to WVURC in the event WVURC commits a material breach and such breach is not cured within sixty (60) days following WVURC’s receipt of such notice. WVURC has the right to terminate, or convert all exclusive licenses to nonexclusive licenses in the event TuHURA fails to make payments due under the agreement within thirty (30) days following notice from WVURC; commits a material breach that is not cured, or capable of being cured, within ninety (90) days after receipt of notice from WVURC; or challenges the validity of any of the WVU Patents before a court or other administrative agency in any jurisdiction. Upon any termination prior to the expiration of the WVU Agreement for any reason, all licenses and rights granted pursuant to the agreement will automatically terminate.
The following is a summary of the patent rights licensed from Moffitt Cancer Center and WVURC:
Patents Under License Agreement with West Virginia University Research Corporation
PCT/US2022/070893 (filed 3/1/2022) – “A Delta-Opioid Receptor Targeted Agent for
Molecular Imaging and Immunotherapy of Cancer”
Applicant: West Virginia University Board of Governors on behalf of West Virginia University
Summary: Relates to molecular conjugates of anticancer compounds and imaging agents and methods of use as a cancer therapy comprising an antagonist of a cell surface opioid receptor such as a delta opioid receptor (DOR), specific to a target cell, an imaging agent, and an immune modulatory molecule, such as a T cell modulator, conjugated to the DOR antagonist.
Earliest Expected Expiration Date: 3/1/2042
| Country |
App. No. |
Filing Date |
Grant Date |
Patent No. |
Status | |||||
| AU |
2022229527 | 8/23/2023 | Pending | |||||||
| CA |
3209499 | 8/23/2023 | Pending | |||||||
| CN |
202280018635.7 | 10/8/2023 | Pending | |||||||
| HK |
62024085712.3 | 1/19/2024 | Pending | |||||||
| EPO |
22764254.3 | 9/28/2023 | Pending | |||||||
| IN |
202317063261 | 9/20/2023 | Pending | |||||||
| JP |
2023-553656 | 8/31/2023 | Pending | |||||||
| KR |
10-2023-7033553 | 9/27/2023 | Pending | |||||||
| US |
18/548724 | 9/1/2023 | Pending |
PCT/US2022/070894 (filed 3/1/2022) – “A Delta-Opioid Receptor Targeted Agent for
Molecular Imaging and Immunotherapy of Cancer”
Applicants: West Virginia University Board of Governors on behalf of West Virginia University and TuHURA Biopharma Inc.
Summary: Relates to molecular conjugates of anticancer compounds and imaging agents and methods of use as a cancer therapy comprising an antagonist of a cell surface opioid receptor such as a DOR or agents that are kinase inhibitors or JAK/STAT3 inhibitors, specific to a target cell, an imaging agent, and an immune modulatory molecule, such as a T cell modulator, conjugated to the DOR antagonist or kinase inhibitors or JAK/STAT3 inhibitors.
Earliest Expected Expiration Date: 3/1/2042
| Country |
App. No. |
Filing Date |
Grant Date |
Patent No. |
Status | |||||
| AU |
2022231182 | 8/23/2023 | Pending | |||||||
| CA |
3210556 | 8/31/2023 | Pending | |||||||
| CN |
202280018634.2 | 10/20/2023 | Pending | |||||||
| EPO |
22764255.0 | 9/28/2023 | Pending | |||||||
| IN |
202317062268 | 9/15/2023 | Pending | |||||||
| JP |
2023-553657 | 8/31/2023 | Pending | |||||||
| KR |
10-2023-7033611 | 9/27/2023 | Pending | |||||||
| US |
18/548729 | 9/1/2023 | Pending |
Patents Under License Agreements with H. Lee Moffitt Cancer Center
PCT/US2017/030962 (filed 5/4/2017) – “A Delta-Opioid Receptor Targeted Agent for
Molecular Imaging and Immunotherapy of Cancer”
Applicants: University of South Florida and the H. Lee Moffitt Cancer Center
Summary: Relates to compounds comprising at least one delta-opioid receptor ligand, such as Dmt-Tic, conjugated to an anti-PDl checkpoint inhibitor antibody and Dmt-Tic-antibody conjugates and methods of use thereof to treat cancer.
Earliest Expected Expiration Date: 5/4/2037
| Country |
App. No. |
Filing Date |
Grant Date |
Patent No. |
Status | |||||
| US |
16/098906 | 11/5/2018 | 10/1/2019 | 10426843 | Granted | |||||
| US |
16/587720 | 9/30/2019 | Inactive | |||||||
| US |
17/830781 | 6/2/2022 | 1/7/2025 | 12186404 | Granted | |||||
| US |
19/011258 | 1/6/2025 | Pending |
PCT/US2015/038057 (filed 6/26/2015) – “Conjugates for Immunotherapy”
Applicants: University of South Florida and the H. Lee Moffitt Cancer Center
Summary: Relates to molecular conjugates comprising agonists of cell surface receptors specific to a target cell, such as DOR agonists, and an immune effector, such as a T cell modulator, compositions comprising the same and methods of treating a disease, such as cancer, by administering the molecular conjugates.
Earliest Expected Expiration Date: 6/26/2035
| Country |
App. No. |
Filing Date |
Grant Date |
Patent No. |
Status | |||||
| US |
15/321316 | 12/22/2016 | 10/22/2019 | 10449227 | Granted | |||||
| US |
16/659207 | 10/21/2019 | Inactive | |||||||
| US |
17/889456 | 8/17/2022 | Inactive | |||||||
| US |
18/130049 | 4/3/2023 | Pending |
PCT/US2021/022464 (filed 3/16/2021) – “Delta opioid receptor antagonists reprogram
immunosuppressive microenvironment to boost immunotherapy”
Applicants: H. Lee Moffitt Cancer Center
Summary: Relates to (a) methods of stimulating endogenous T cells, increasing the efficacy of adoptive immunotherapy, or reprogramming immunosuppressive tumor microenvironments, immunosuppressive myelopoiesis, or myeloid-derived suppressor cells by administering a DOR antagonist; (b) combination immunotherapies comprising an adoptive immunotherapy or an immune system activator and a DOR antagonist and methods of using the same to treat cancer; (c) methods of treating autoimmune disease or microbial infection by administering a DOR agonist, optionally with an immunosuppressor; and (d) combination therapies comprising a DOR agonist and an immunosuppressor.
Earliest Expected Expiration Date: 3/16/2041
| Country |
App. No. |
Filing Date |
Grant Date |
Patent No. |
Status | |||||
| US | 17/912300 | 9/16/2022 | Pending |
Employees and Human Capital Resources
As of September 11, 2025, TuHURA had 22 full-time employees and no part-time employees. Of these employees, 18 were engaged in research and development activities. The majority of TuHURA’s employees are based in Tampa, Florida. None of TuHURA’s employees are represented by labor unions or covered by collective bargaining agreements. TuHURA considers its relationship with its employees to be good.
TuHURA’s human capital resources objectives include, as applicable, identifying, recruiting, retaining, incentivizing and integrating TuHURA’s existing and new employees, advisors and consultants. The principal purposes of TuHURA’s equity and cash incentive plans are to attract, retain and reward personnel through the granting of stock-based and cash-based compensation awards, in order to increase stockholder value and the success of the company by motivating such individuals to perform to the best of their abilities and achieve its objectives.
Facilities
TuHURA’s principal office is located in Tampa, Florida. TuHURA currently leases approximately 12,199 square feet of office and laboratory space under a lease that is due to expire in March 2026. TuHURA believes that such office and laboratory space will be sufficient for TuHURA’s planned operations for the foreseeable future.
Government Regulation and Product Approval
Therapeutic products are subject to rigorous regulation by the FDA and other governmental agency regulations in the United States and in foreign countries. Noncompliance with applicable requirements can result in import detentions, fines, civil monetary penalties, injunctions, suspensions or losses of regulatory approvals or licenses, recall or seizure of products, operating restrictions, denial of export applications, governmental prohibitions on entering into supply contracts, and criminal penalties and prosecution. Failure to obtain regulatory approvals or the restriction, suspension or revocation of regulatory approvals or licenses, as well as any other failure to comply with regulatory requirements, would have a material adverse effect on TuHURA’s business, financial condition and results of operations. In connection with seeking therapeutic approval, TuHURA will have to comply with the many regulations and requirements associated with the conduct of preclinical and clinical trials, the FDA application process, the terms of any pre-certification protocols and agreements, FDA manufacturing requirements for investigational products, and testing. Upon approval of a Biologics License Application, or BLA, and similar approvals in other jurisdictions, there will be additional regulations that must be complied with, including regulations relating to the packaging, distribution, marking, marketing and claims of TuHURA’s potential products. These later regulations are not only found in federal regulation but many states and, of course, foreign countries.
The U.S. FDA Process
The FDA regulates the clinical testing and design of therapeutics to ensure that medical products distributed in the United States are safe and effective for their intended uses. The application process for a new therapeutic is highly regulated.
As a biopharmaceutical company that operates in the United States, TuHURA is subject to extensive regulation by relevant authorities, including the FDA. TuHURA’s potential products will be regulated as biologics. With this classification, commercial production of its potential products will need to occur in registered and licensed facilities in compliance with current good manufacturing practices (cGMP) established by the FDA for biologics. The FDA categorizes human cell- or tissue-based products as either minimally manipulated or more than minimally manipulated, and has determined that more than minimally manipulated products require clinical trials to demonstrate product safety and efficacy and the submission of a BLA for marketing authorization.
Government authorities in the United States (at the federal, state and local levels) and in other countries extensively regulate, among other things, the research, development, testing, manufacturing, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, post-approval monitoring and reporting, marketing and export and import of biopharmaceutical products such as those TuHURA is developing. TuHURA’s candidates must be approved by the FDA before they may be legally marketed in the United States and by the appropriate foreign regulatory agency before they may be legally marketed in a foreign country. Generally, TuHURA’s activities in other countries will be subject to regulation that is similar in nature and scope as that imposed in the United States, although there can be important differences. Additionally, some significant aspects of regulation in Europe are addressed in a centralized way, but country-specific regulation remains essential in many respects. The process for obtaining regulatory marketing approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
U.S. Product Development Process
In the United States, the FDA regulates pharmaceutical and biological products under the Federal Food, Drug, and Cosmetic Act, or FDCA, the Public Health Service Act, or PHSA, and their respective implementing regulations. Products are also subject to other federal, state and local statutes and regulations. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. FDA sanctions could include, among other actions, refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product recalls or withdrawals from the market, product seizures, total or partial suspension of production or distribution injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. Any agency or judicial enforcement action could have a material adverse effect on TuHURA. The FDA has limited experience with commercial development of T cell therapies for cancer, including direct- injectable technologies such as AIM INJ. The process required by the FDA before a biological product may be marketed in the United States generally involves the following:
| • | completion of nonclinical laboratory tests and animal studies according to Good Laboratory Practices, or GLPs, and applicable requirements for the humane use of laboratory animals or other applicable regulations; |
| • | submission to the FDA of an investigational new drug, or IND, application, which must become effective before human clinical trials may begin; |
| • | approval by an independent institutional review board, or IRB, or ethics committee at each clinical trial site before each clinical trial may be initiated; |
| • | performance of adequate and well-controlled human clinical trials according to the FDA’s regulations commonly referred to as Good Clinical Practice, or GCP, and any additional requirements for the protection of human research patients and their health information, to establish the safety and efficacy of the proposed biological product for its intended use; |
| • | submission to the FDA of a BLA for marketing approval that includes substantive evidence of safety, purity, and potency from results of nonclinical testing and clinical trials; |
| • | satisfactory completion of an FDA inspection of the manufacturing facility or facilities where the biological product is produced to assess compliance with cGMP to assure that the facilities, methods and controls are adequate to preserve the biological product’s identity, strength, quality and purity and, if applicable, the FDA’s current Good Tissue Practices, or cGTPs, for the use of human cellular and tissue products; |
| • | potential FDA audit of the trial and clinical trial sites that generated the data in support of the BLA; and |
| • | FDA review and approval, or licensure, of the BLA. |
Preclinical studies
Before testing any biological product candidate, including TuHURA’s drug candidates, in humans, the drug candidate enters the preclinical testing stage. Preclinical tests, also referred to as nonclinical studies, include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the drug candidate. The conduct of the preclinical tests must comply with federal regulations and requirements, including GLP. The clinical trial sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. Some preclinical testing may continue even after the IND is submitted. An IND is a request for authorization from the FDA to administer an investigational product to humans, and must become effective before human clinical trials may begin.
Human clinical trials in support of a BLA
Clinical trials involve the administration of the biological product candidate to human research subjects under the supervision of qualified investigators, generally licensed physicians not employed by or under the trial sponsor’s control. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, dosing procedures, subject selection, inclusion and exclusion criteria, and the parameters to be used to monitor subject safety, including stopping rules that assure a clinical trial will be stopped if certain adverse events should occur. Each protocol and any amendments to the protocol must be submitted to the FDA as part of the IND. An IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA raises concerns or questions regarding the proposed clinical trials and places the trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. The FDA may also impose clinical holds on a biological product candidate at any time before or during a clinical trial due to safety concerns or non-compliance. If the FDA imposes a clinical hold, the trial may not recommence without FDA authorization and then only under terms authorized by the FDA. Accordingly, TuHURA cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin or that, once begun, issues will not arise that suspend or terminate such trials.
Clinical trials must be conducted and monitored in accordance with the FDA’s regulations comprising the GCP requirements, including the requirement that all research participants provide informed consent. Further, each clinical trial must be reviewed and approved by an independent institutional review board, or IRB, at or servicing each institution at which the clinical trial will be conducted. An IRB is charged with protecting the welfare and rights of trial participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the form and content of the informed consent form that must be signed by each clinical trial subject or the participant’s legal representative and must monitor the clinical trial until completed. For certain clinical trials involving biologics, they also must be reviewed by an institutional biosafety committee, or IBC, a local institutional committee that reviews and oversees basic and clinical research conducted at that institution. The IBC assesses the safety of the research and identifies any potential risk to public health or the environment.
Information about certain clinical trials, including details of the protocol and eventually study results, also must be submitted within specific timeframes to the National Institutes of Health, or NIH, for public dissemination on the ClinicalTrials.gov data registry. Information related to the investigational product, patient population, phase of investigation, study sites and investigators and other aspects of the clinical trial is made public as part of the registration of the clinical trial. Sponsors are also obligated to disclose the results of their clinical trials after completion. Disclosure of the results of these trials can be delayed in some cases for up to two years after the date of completion of the trial.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| • | phase 1. The investigational biological product candidate is initially introduced into human subjects to test for safety, dosage tolerance, absorption, metabolism, distribution and excretion. The initial human testing is often conducted in patients, rather than in healthy volunteers, in the case of products for severe or life-threatening diseases. |
| • | phase 2. The biological product is evaluated in a limited patient population to identify possible safety risks (adverse effects), optimize dosing and preliminarily evaluate the efficacy of the product for specific targeted diseases. |
| • | phase 3. Clinical trials are undertaken in an expanded patient population to further evaluate dosage, clinical efficacy, and safety, often at geographically dispersed trial sites. These clinical trials are intended to establish the overall risk to benefit ratio of the investigational product and provide, if appropriate, an adequate basis for product labeling. These trials may include comparisons with placebo and/or other comparator treatments. The duration of treatment is often extended to mimic the actual use of a product during marketing. |
Post-approval clinical trials, sometimes referred to as phase 4 clinical trials, may be conducted after initial marketing approval. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication, particularly for long-term safety follow-up. In certain instances, the FDA may mandate the performance of phase 4 clinical trials as a condition of approval of a BLA.
Progress reports detailing the results of the clinical trial must be submitted at least annually to the FDA and more frequently if serious adverse events, or SAEs, occur. The FDA or the trial sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research participants are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the clinical protocol, GCP, or other IRB requirements, or if the investigational product has been associated with unexpected serious harm to patients. Additionally, some trials are overseen by an independent group of qualified experts organized by the trial sponsor known as the data safety monitoring board or committee. This group provides authorization for whether a trial may move forward at designated checkpoints.
During the development of a new drug or biological product, sponsors have the opportunity to meet with the FDA at certain points, including prior to submission of an IND, at the end of phase 2, and before submission of a BLA. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date, for the FDA to provide guidance, and for the sponsor and the FDA to reach agreement on the next phase of development. Sponsors typically use the end of phase 2 meeting to discuss their phase 2 clinical results with the agency and to present their plans for the pivotal phase 3 studies that they believe will support approval of the new drug or biological product.
Human immunotherapy products are a new category of therapeutics. Because this is a relatively new and expanding area of novel therapeutic interventions, there can be no assurance as to the length of the clinical trial period, the number of participants the FDA will require to be enrolled in the trials in order to establish the safety, efficacy, purity and potency of immunotherapy products, or that the data generated in these trials will be acceptable to the FDA to support marketing approval.
Concurrently with clinical trials, companies usually complete additional studies and must also develop additional information about the physical characteristics of the biological product as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. To help reduce the risk of the introduction of adventitious agents with use of biological products, the PHSA emphasizes the importance of manufacturing control for products whose attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the drug candidate and, among other things, the sponsor must develop methods for testing the identity, strength, quality, potency and purity of the final biological product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the biological drug candidate does not undergo unacceptable deterioration over its shelf life.
U.S. Review and Approval Processes
Assuming successful completion of the required clinical testing, the results of the preclinical studies and clinical trials, along with information relating to the product’s chemistry, manufacturing, and controls and proposed labeling, are submitted to the FDA as part of a BLA requesting approval to market the product for one or more indications. A BLA in particular must contain proof of the biological product candidate’s safety, purity, potency and efficacy for its proposed indication or indications. Data may come from company-sponsored clinical trials intended to test the safety and efficacy of a product’s use or from a number of alternative sources, including studies initiated by investigators. To support marketing approval, the data submitted must be sufficient in quality and quantity to establish the safety and efficacy of the investigational product to the satisfaction of the FDA. The testing and approval processes require substantial time and effort and there can be no assurance that the FDA will accept the BLA for filing and, even if filed, that any approval will be granted on a timely basis, if at all.
Under the Prescription Drug User Fee Act, as amended, or PDUFA, each BLA must be accompanied by a significant user fee, and the sponsor of an approved BLA is also subject to an annual program fee. The FDA adjusts the PDUFA user fees on an annual basis. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on BLAs for products designated as orphan drugs, unless the product also includes a non- orphan indication.
According to the goals and policies for original BLAs agreed to by the FDA under PDUFA, the FDA has ten months from the accepted for filing date in which to complete its initial review of a standard application and respond to the applicant, and six months from the filing date for an application with priority review. For all BLAs, the ten and six-month time periods run from the filing date; for most other original applications, the ten and six-month time periods run from the submission date. Despite these review goals, it is not uncommon for FDA review of a BLA to extend beyond the goal date.
Within 60 days following submission of the application, the FDA reviews a BLA submitted to determine if it is substantially complete before the agency accepts it for filing. The FDA may refuse to file any BLA that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the BLA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review of the BLA. The FDA reviews the BLA to determine, among other things, whether the proposed product is safe, potent, and/or effective for its intended use, and has an acceptable purity profile, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, safety, strength, quality, potency and purity. The review process may be extended by the FDA for three additional months to consider new information or in the case of a clarification provided by the applicant to address an outstanding deficiency identified by the FDA following the original submission.
The FDA may refer applications for novel biological products or biological products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making final decisions on approval. The FDA likely will re-analyze the clinical trial data, which could result in extensive discussions between the FDA and the applicant during the review process. The FDA also may require submission of a risk evaluation and mitigation strategy, or REMS, if it determines that a REMS is necessary to ensure that the benefits of the drug outweigh its risks and to assure the safe use of the drug or biological product. The REMS could include medication guides, physician communication plans, assessment plans and/or elements to assure safe use, such as restricted distribution methods, patient registries or other risk minimization tools. The FDA determines the requirement for a REMS, as well as the specific REMS provisions, on a case-by-case basis. If the FDA concludes a REMS is needed, the sponsor of the BLA must submit a proposed REMS. The FDA will not approve a BLA without a REMS, if required.
Before approving a BLA, the FDA will typically conduct a pre-approval inspection of the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. For immunotherapy products, the FDA also will not approve the product if the manufacturer is not in compliance with the cGTPs, to the extent applicable. These are FDA regulations and guidance documents that govern the methods used in, and the facilities and controls used for, the manufacture of human cells, tissues, and cellular and tissue based products (HCT/Ps), which are human cells or tissue intended for implantation, transplant, infusion, or transfer into a human recipient. The primary intent of the cGTP requirements is to ensure that cellular tissue-based products are manufactured in a manner designed to prevent the introduction, transmission and spread of communicable disease. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure that the clinical trials were conducted in compliance with IND trial requirements and GCP requirements. To assure cGMP, cGTP and GCP compliance, an applicant must incur significant expenditure of time, money and effort in the areas of training, record keeping, production, and quality control.
In addition, under the Pediatric Research Equity Act, or PREA, a BLA or supplement to a BLA must contain data to assess the safety and effectiveness of the product for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of data or full or partial waivers. A sponsor who is planning to submit a marketing application for a product that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration is required to submit an initial Pediatric Study Plan, or iPSP, within sixty days of an end-of-phase 2 meeting or, if there is no such meeting, as early as practicable before the initiation of the phase 3 or phase 2/3 clinical trial. The iPSP must include an outline of the pediatric study or studies that the sponsor plans to conduct, including trial objectives and design, age groups, relevant endpoints and statistical approach, or a justification for not including such detailed information, and any request for a deferral of pediatric assessments or a full or partial waiver of the requirement to provide data from pediatric studies along with supporting information. The FDA and the sponsor must reach an agreement on the iPSP. A sponsor can submit amendments to an agreed upon iPSP at any time if changes to the pediatric plan need to be considered based on data collected from preclinical studies, early phase clinical trials or other clinical development programs. Unless otherwise required by regulation, the PREA does not apply to any product for an indication for which orphan designation has been granted. However, if only one indication for a product has orphan designation, a pediatric assessment may still be required for any applications to market that same product for the non-orphan indication(s).
Notwithstanding the submission of relevant data and information, the FDA may ultimately decide that the BLA does not satisfy its regulatory criteria for approval and deny approval or may require additional clinical or other data and information. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than TuHURA interprets the same data. On the basis of the FDA’s evaluation of the BLA and accompanying information, including the results of the inspection of the manufacturing facilities, the FDA may issue either an approval letter or a Complete Response Letter, or CRL. An approval letter authorizes
commercial marketing of the product with specific prescribing information for specific indications. A CRL indicates that the review cycle of the application is complete and the application will not be approved in its present form. A CRL generally outlines the deficiencies in the submission and may require substantial additional testing or information in order for the FDA to reconsider the application. The CRL may require additional clinical or other data, additional pivotal phase 3 clinical trial(s) and/or other significant and time-consuming requirements related to clinical trials, preclinical studies or manufacturing. If a CRL is issued, the applicant may choose to either resubmit the BLA addressing all of the deficiencies identified in the letter, or withdraw the application. If and when those deficiencies have been addressed to the FDA’s satisfaction in a resubmission of the BLA, the FDA will issue an approval letter. The FDA has committed to reviewing such resubmissions in response to an issued CRL in either two or six months depending on the type of information included. Even with the submission of this additional information, however, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
If a product receives regulatory approval from the FDA, the approval is limited to the conditions of use (e.g., patient population, indication) described in the application. Further, depending on the specific risk(s) to be addressed, the FDA may require that contraindications, warnings or precautions be included in the product labeling, require that post-approval trials, including phase 4 clinical trials, be conducted to further assess a product’s safety after approval, require testing and surveillance programs to monitor the product after commercialization, or impose other conditions, including distribution and use restrictions or other risk management mechanisms under a REMS, which can materially affect the potential market and profitability of the product. The FDA may prevent or limit further marketing of a product based on the results of post-marketing trials or surveillance programs. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further testing requirements and FDA review and approval.
Fast Track, Breakthrough Therapy and Priority Review Designations
The FDA is authorized to designate certain products for expedited development or review if they are intended to address an unmet medical need in the treatment of a serious or life-threatening disease or condition. These programs include fast track designation, Breakthrough Therapy Designation and priority review designation and regenerative medicine advanced therapy designation.
To be eligible for a fast track designation, the FDA must determine, based on the request of a sponsor, that a product is intended to treat a serious or life-threatening disease or condition and demonstrates the potential to address an unmet medical need by providing a therapy where none exists or a therapy that may be potentially superior to existing therapy based on efficacy or safety factors. Fast track designation provides opportunities for more frequent interactions with the FDA review team to expedite development and review of the product. The FDA may also review sections of the BLA for a fast track product on a rolling basis before the complete application is submitted, if the sponsor and the FDA agree on a schedule for the submission of the application sections and the sponsor pays any required user fees upon submission of the first section of the NDA or BLA. In addition, fast track designation may be withdrawn by the sponsor or rescinded by the FDA if the designation is no longer supported by data emerging from the clinical trial process.
In addition, with the enactment of the Food and Drug Administration Safety and Innovation Act, or FDASIA, in 2012, Congress created a new regulatory program for product candidates designated by FDA as “breakthrough therapies” upon a request made by the IND sponsors. A breakthrough therapy is defined as a drug or biologic that is intended, alone or in combination with one or more other drugs or biologics, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug or biologic may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Drugs or biologics designated as breakthrough therapies are also eligible for accelerated approval of their respective marketing applications. The FDA must take certain actions with respect to breakthrough therapies, such as holding timely meetings with and providing advice to the product sponsor, which are intended to expedite the development and review of an application for approval of a breakthrough therapy.
Next, the FDA may designate a product for priority review if it is a drug or biologic that treats a serious condition and, if approved, would provide a significant improvement in safety or effectiveness. The FDA determines at the time that the marketing application is submitted, on a case-by-case basis, whether the proposed drug represents a significant improvement in treatment, prevention or diagnosis of disease when compared with other available therapies. Significant improvement may be illustrated by evidence of increased effectiveness in the treatment of a condition, elimination or substantial reduction of a treatment-limiting drug reaction, documented enhancement of patient compliance that may lead to improvement in serious outcomes, or evidence of safety and effectiveness in a new subpopulation. A priority review designation is intended to direct overall attention and resources to the evaluation of such applications, and to shorten the FDA’s goal for taking action on a marketing application from ten months to six months for an original BLA from the date of filing.
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened. Furthermore, fast track designation, breakthrough therapy designation and priority review do not change the standards for approval and may not ultimately expedite the development or approval process.
As part of the 21st Century Cures Act, congress created an accelerated approval pathway for regenerative medicine advanced therapies, or RMATs, which includes therapeutic tissue engineered products, human cell and tissue products, cell therapies and combination products using any such therapies. The program is intended to facilitate expedited development and review of RMATs intended to address serious diseases or conditions.
A sponsor may request a RMAT designation from the FDA concurrently with or any time after the IND submission. The FDA has 60 calendar days to determine if the drug product meets the required criteria. Preliminary clinical evidence that the product has the potential to address a serious unmet need or condition is expected, is not required to indicate that the drug product may offer significant improvement over current therapies. The RMAT designation provides the same benefits of the fast track and breakthrough designation programs and programs may be eligible for priority review. Products with the RMAT designation may also be eligible for accelerated approval if pre-agreed criteria are met.
Accelerated Approval Pathway
In addition, products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval from the FDA and may be approved on the basis of adequate and well-controlled clinical trials establishing that the drug product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit. The FDA may also grant accelerated approval for such a drug or biologic when the product has an effect on an intermediate clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality, or IMM, and that is reasonably likely to predict an effect on IMM or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA may require that a sponsor of a drug receiving accelerated approval perform post-marketing clinical trials to verify and describe the predicted effect on IMM or other clinical endpoint, and the product may be subject to expedited withdrawal procedures. Drugs and biologics granted accelerated approval must meet the same statutory standards for safety and effectiveness as those granted traditional approval.
For the purposes of accelerated approval, a surrogate endpoint is a marker, such as a laboratory measurement, radiographic image, physical sign, or other measure that is thought to predict clinical benefit, but is not itself a measure of clinical benefit. Surrogate endpoints can often be measured more easily or more rapidly than clinical endpoints. An intermediate clinical endpoint is a measurement of a therapeutic effect that is considered reasonably likely to predict the clinical benefit of a drug, such as an effect on IMM. The FDA has limited experience with accelerated approvals based on intermediate clinical endpoints, but has indicated that such endpoints generally may support accelerated approval when the therapeutic effect measured by the endpoint is not itself a clinical benefit and basis for traditional approval, if there is a basis for concluding that the therapeutic effect is reasonably likely to predict the ultimate long-term clinical benefit of a drug.
The accelerated approval pathway is most often used in settings in which the course of a disease is long and an extended period of time is required to measure the intended clinical benefit of a drug, even if the effect on the surrogate or intermediate clinical endpoint occurs rapidly. For example, accelerated approval has been used extensively in the development and approval of drugs for treatment of a variety of cancers in which the goal of therapy is generally to improve survival or decrease morbidity and the duration of the typical disease course requires lengthy and sometimes large clinical trials to demonstrate a clinical or survival benefit.
The accelerated approval pathway is usually contingent on a sponsor’s agreement to conduct, in a diligent manner, additional post-approval confirmatory studies to verify and describe the drug’s clinical benefit. As a result, a product candidate approved on this basis is subject to rigorous post-marketing compliance requirements, including the completion of phase 4 or post-approval clinical trials to confirm the effect on the clinical endpoint. Failure to conduct required post-approval studies, or to confirm the predicted clinical benefit of the product during post-marketing studies, would allow the FDA to withdraw approval of the drug. All promotional materials for product candidates being considered and approved under the accelerated approval program are subject to prior review by the FDA.
Orphan Drug Designation and Exclusivity
Under the Orphan Drug Act, the FDA may grant orphan drug designation to a drug or biologic product intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that the cost of developing and making available in the United States a drug or biologic for this type of disease or condition will be recovered from sales in the United States for that drug or biologic. Orphan drug designation must be requested before submitting an NDA or BLA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use will be disclosed publicly by the FDA; the posting will also indicate whether the drug or biologic is no longer designated as an orphan drug. More than one product candidate may receive an orphan drug designation for the same indication. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to seven years of orphan product exclusivity. During the seven-year exclusivity period, the FDA may not approve any other applications to market a product containing the same active moiety for the same disease, except in very limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity. A product is clinically superior if it is safer, more effective or makes a major contribution to patient care. Thus, orphan drug exclusivity could block the approval of one of TuHURA’s potential products for seven years if a competitor obtains approval of the same product as defined by the FDA and TuHURA is not able to show the clinical superiority of its product candidate or if its product candidate’s indication is determined to be contained within the competitor’s product orphan indication. In addition, the FDA will not recognize orphan drug exclusivity if a sponsor fails to demonstrate upon approval that the product is clinically superior to a previously approved product containing the same active moiety for the same orphan condition, regardless of whether or not the approved product was designated an orphan drug or had orphan drug exclusivity.
Patent Term Restoration
Depending upon the timing, duration and specifics of FDA approval of TuHURA’s biological products, some of TuHURA’s US patents may be eligible for limited patent term extension. These patent term extensions permit a patent restoration term of up to five years as compensation for any patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between the effective date of an IND, and the submission date of a BLA, plus the time between the submission date of a BLA and the approval of that application. Only one patent applicable to an approved biological product is eligible for the extension, and the extension must be applied for prior to expiration of the patent. The United States Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration.
Pediatric Exclusivity
Pediatric exclusivity is a type of non-patent marketing exclusivity available in the United States and, if granted, it provides for the attachment of an additional six months of marketing protection to the term of any existing regulatory exclusivity or listed patents. This six-month exclusivity may be granted if a sponsor submits pediatric data that fairly respond to a written request from the FDA for such data. The data do not need to show the product to be effective in the pediatric population studied; rather, if the clinical trial is deemed to fairly respond to the FDA’s request, the additional protection is granted. If reports of requested pediatric studies are submitted to and accepted by the FDA within the statutory time limits, whatever statutory or regulatory periods of exclusivity or patent protection cover the product are extended by six months. This is not a patent term extension, but it effectively extends the regulatory period during which the FDA cannot approve another application. The issuance of a Written Request does not require the sponsor to undertake the described studies.
Reference Product Exclusivity for Biological Products
In March 2010, the Patient Protection and Affordable Care Act was enacted in the United States and included the Biologics Price Competition and Innovation Act of 2009, or the BPCIA. The BPCIA amended the PHSA to create an abbreviated approval pathway for biological products that are biosimilar to or interchangeable with an FDA-licensed reference biological product. To date, the FDA has approved a number of biosimilars, and numerous biosimilars have been approved in Europe. The FDA has also issued several guidance documents outlining its approach to reviewing and approving biosimilars and interchangeable biosimilars.
A biosimilar product is defined as one that is highly similar to a reference product notwithstanding minor differences in clinically inactive components and for which there are no clinically meaningful differences between the biological product and the reference product in terms of the safety, purity and potency of the product. An interchangeable product is a biosimilar product that can be expected to produce the same clinical results as the reference product in any given patient and, for products administered multiple times to an individual, that the product and the reference product may be alternated or switched after one has been previously administered without increasing safety risks or risks of diminished efficacy relative to exclusive use of the reference biological product without such alternation or switch. Upon licensure by the FDA, an interchangeable biosimilar may be substituted for the reference product without the intervention of the health care provider who prescribed the reference product.
The biosimilar applicant must demonstrate that the product is biosimilar based on data from (i) analytical studies showing that the biosimilar product is highly similar to the reference product; (ii) animal studies (including toxicity); and (iii) one or more clinical studies to demonstrate safety, purity and potency in one or more appropriate conditions of use for which the reference product is approved. In addition, the applicant must show that the biosimilar and reference products have the same mechanism of action for the conditions of use on the label, route of administration, dosage and strength, and the production facility must meet standards designed to assure product safety, purity and potency.
A reference biological product is granted 12 years of regulatory exclusivity from the time of first licensure of the product, and the first approved interchangeable biologic product to a reference product will be granted an exclusivity period of up to one year after it is first commercially marketed. If pediatric studies are performed and accepted by the FDA as responsive to a Written Request, the 12-year exclusivity period will be extended for an additional six months. In addition, the FDA will not accept an application for a biosimilar or interchangeable product based on the reference biological product until four years after the date of first licensure of the reference product. “First licensure” typically means the initial date the particular product at issue was licensed in the United States. Date of first licensure does not include the date of licensure of (and a new period of exclusivity is not available for) a supplement for the reference product for a subsequent application filed by the same sponsor or manufacturer of the reference product (or licensor, predecessor in interest or other related entity) for a change (not including a modification to the structure of the biological product) that results in a new indication, route of administration, dosing schedule, dosage form, delivery system, delivery device or strength or for a modification to the structure of the biological product that does not result in a change in safety, purity or potency. Therefore, one must determine whether a new product includes a modification to the structure of a previously licensed product that results in a change in safety, purity or potency to assess whether the licensure of the new product is a first licensure that triggers its own period of exclusivity. Whether a subsequent application, if approved, warrants exclusivity as the “first licensure” of a biological product is determined on a case-by-case basis with data submitted by the sponsor.
The BPCIA is complex and is still being interpreted and implemented by the FDA. In addition, recent government proposals have sought to reduce the 12-year reference product exclusivity period. Other aspects of the BPCIA, some of which may impact the BPCIA exclusivity provisions, have also been the subject of recent litigation. As a result, the ultimate impact, implementation and meaning of the BPCIA is subject to continued uncertainty.
Post-Approval Requirements
Any potential products for which TuHURA receives FDA approvals are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, and complying with FDA promotion and advertising requirements, which include, among others, standards for direct-to-consumer advertising, restrictions on promoting products for uses or in patient populations that are not described in the product’s approved uses (known as off-label use), limitations on industry-sponsored scientific and educational activities, and requirements for promotional activities involving the internet. Although physicians may prescribe legally available products for off-label uses, if the physicians deem to be appropriate in their professional medical judgment, it is FDA’s position that manufacturers may not market or promote such off-label uses. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including liability under federal fraud and abuse and civil and criminal false claims laws. If there are any modifications to the product, including changes in indications, labeling or manufacturing processes or facilities, the applicant may be required to submit and obtain FDA approval of a new BLA or a supplement, which may require the applicant to develop additional data or conduct additional preclinical studies and clinical trials. The FDA may also place other conditions on approvals including the requirement for a REMS to assure the safe use of the product. A REMS could include medication guides, physician communication plans or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. Any of these limitations on approval or marketing could restrict the commercial promotion, distribution, prescription or dispensing of products. Product approvals may be withdrawn for non-compliance with regulatory standards or if problems occur following initial marketing.
In addition, quality control and manufacturing procedures must continue to conform to applicable manufacturing requirements after approval to ensure the quality and long-term stability of the product. TuHURA expects to rely on third parties for the production of clinical and commercial quantities of TuHURA’s potential products in accordance with cGMP regulations. The cGMP regulations include requirements relating to organization of personnel, buildings and facilities, equipment, control of components and drug product containers and closures, production and process controls, packaging and labeling controls, holding and distribution, laboratory controls, records and reports and returned or salvaged products. The manufacturing facilities for TuHURA’s product candidates must meet cGMP requirements and satisfy the FDA or comparable foreign regulatory authorities before any product is approved and TuHURA’s commercial products can be manufactured. TuHURA relies, and expects to continue to rely, on third parties for the production of clinical and commercial quantities of TuHURA’s products in accordance with cGMP regulations. These manufacturers must comply with cGMP regulations that require, among other things, quality control and quality assurance, the maintenance of records and documentation and the obligation to investigate and correct any deviations from cGMP. Manufacturers and other entities involved in the manufacture and distribution of approved products are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. Accordingly, manufacturers must continue to expend time, money, and effort in the area of production and quality control to maintain cGMP compliance. Future inspections by the FDA and other regulatory agencies may identify compliance issues at the facilities of TuHURA’s contract manufacturing organizations, or CMOs, that may disrupt production or distribution or require substantial resources to correct. In addition, the discovery of conditions that violate these rules, including failure to conform to cGMP regulations, could result in enforcement actions, and the discovery of problems with a product after approval may result in restrictions on a product, manufacturer, or holder of an approved BLA, including, among other things, voluntary recall and regulatory sanctions as described below.
Once an approval of a product is granted, the FDA may withdraw the approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in mandatory revisions to the approved labeling to add new safety information; imposition of post-market or clinical trials to assess new safety risks; or imposition of distribution or other restrictions under a REMS program. Other potential consequences include, among other things:
| • | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| • | fines, warning letters or other enforcement-related letters or clinical holds on post-approval clinical trials; |
| • | refusal of the FDA to approve pending BLAs or supplements to approved BLAs, or suspension or revocation of product approvals; |
| • | product seizure or detention, or refusal to permit the import or export of products; |
| • | injunctions or the imposition of civil or criminal penalties; and |
| • | consent decrees, corporate integrity agreements, debarment, or exclusion from federal health care programs; or mandated modification of promotional materials and labeling and the issuance of corrective information. |
In addition, the Drug Supply Chain Security Act, or DSCSA, was enacted with the aim of building an electronic system to identify and trace certain prescription drugs distributed in the United States, including most biological products. The DSCSA mandates phased-in and resource-intensive obligations for pharmaceutical manufacturers, wholesale distributors, and dispensers over a 10-year period that has been extended an additional year to be implemented in November 2024. In the Fall of 2024, the FDA granted an additional extension to 2025 based on the type of activities being performed. From time to time, new legislation and regulations may be implemented that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. It is impossible to predict whether further legislative or regulatory changes will be enacted, or FDA regulations, guidance or interpretations changed or what the impact of such changes, if any, may be.
Regulation Outside of the United States
In addition to regulations within the United States, TuHURA will be subject to a variety of foreign regulations governing clinical trials and the commercial sale and distribution of TuHURA’s products outside of the United States. Whether or not TuHURA obtains FDA approval for a product candidate, TuHURA must obtain approval by the comparable regulatory authorities of foreign countries or economic areas, such as the 27- member European Union, before TuHURA may commence clinical trials or market products in those countries or areas. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly between countries and jurisdictions and can involve additional testing and additional administrative review periods. The time required to obtain approval in other countries and jurisdictions might differ from and be longer than that required to obtain FDA approval. Regulatory approval in one country or jurisdiction does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country or jurisdiction may negatively impact the regulatory process in others.
European Union drug development, review and approval
In the European Union, TuHURA’s product candidates also may be subject to extensive regulatory requirements. As in the United States, medicinal products can be marketed only if a marketing authorization from the competent regulatory agencies has been obtained. Similar to the United States, the various phases of preclinical and clinical research in the European Union are subject to significant regulatory controls.
The Clinical Trials Directive 2001/20/EC, the Directive 2005/28/EC on GCP, and the related national implementing provisions of the individual EU Member States govern the system for the approval of clinical trials in the European Union. Under this system, an applicant must obtain prior approval from the competent national authority of the EU Member States in which the clinical trial is to be conducted. Furthermore, the applicant may only start a clinical trial at a specific study site after the competent ethics committee has issued a favorable opinion. The clinical trial application must be accompanied by, among other documents, an IMPD (the Common Technical Document) with supporting information prescribed by Directive.
2001/20/EC, Directive 2005/28/EC, where relevant the implementing national provisions of the individual EU Member States and further detailed in applicable guidance documents. All suspected unexpected serious adverse reactions to the investigated drug that occur during the clinical trial have to be reported to the competent national authority and the Ethics Committee of the Member State where they occurred.
In April 2014, the new Clinical Trials Regulation, (EU) No 536/2014 (Clinical Trials Regulation) was adopted and came into application in January 2022. The Clinical Trials Regulation is directly applicable in all the EU Member States, repealing the prior Clinical Trials Directive 2001/20/EC.
The new Clinical Trials Regulation simplifies and streamlines the approval of clinical trials in the European Union. The main characteristics of the regulation include: a streamlined application procedure via a single entry point, the “EU portal”; a single set of documents to be prepared and submitted for the application as well as simplified reporting procedures for clinical trial sponsors; and a harmonized procedure for the assessment of applications for clinical trials, which is divided in two parts. Part I is assessed by the competent authorities of all EU Member States in which an application for authorization of a clinical trial has been submitted (Member States concerned). Part II is assessed separately by each Member State concerned. Strict deadlines have been established for the assessment of clinical trial applications. The role of the relevant ethics committees in the assessment procedure will continue to be governed by the national law of the concerned EU Member State. However, overall related timelines will be defined by the Clinical Trials Regulation.
To obtain a marketing authorization of a drug in the European Union, TuHURA may submit marketing authorization applications, or MAA, either under the so-called centralized or national authorization procedures.
Centralized Procedure
The centralized procedure provides for the grant of a single marketing authorization following a favorable opinion by the European Medicines Agency, or EMA, that is valid in all 27 European Union member states, or EU member states, as well as Iceland, Liechtenstein and Norway. The centralized procedure is compulsory for medicines produced by specified biotechnological processes, products designated as orphan medicinal products, advanced-therapy medicines (such as gene-therapy, somatic cell-therapy or tissue-engineered medicines) and products with a new active substance indicated for the treatment of specified diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative disorders or autoimmune diseases and other immune dysfunctions and viral diseases. The centralized procedure is optional for products that represent a significant therapeutic, scientific or technical innovation, or whose authorization would be in the interest of public health. Under the centralized procedure the maximum timeframe for the evaluation of an MAA by the EMA is 210 days, excluding clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the Committee for Medicinal Products for Human Use, or the CHMP. Accelerated assessment might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of a major public health interest, particularly from the point of view of therapeutic innovation. The timeframe for the evaluation of an MAA under the accelerated assessment procedure is of 150 days, excluding stop-clocks.
National authorization procedures
There are also two other possible routes to authorize medicinal products in several EU countries, which are available for investigational medicinal products that fall outside the scope of the centralized procedure:
| • | Decentralized procedure. Using the decentralized procedure, an applicant may apply for simultaneous authorization in more than one EU country of medicinal products that have not yet been authorized in any EU country and that do not fall within the mandatory scope of the centralized procedure. |
| • | Mutual recognition procedure. In the mutual recognition procedure, a medicine is first authorized in one EU Member State, in accordance with the national procedures of that country. Following this, further marketing authorizations can be sought from other EU countries in a procedure whereby the countries concerned agree to recognize the validity of the original, national marketing authorization. |
Under the above-described procedures, before granting the marketing authorization, the EMA or the competent authorities of the Member States of the EEA make an assessment of the risk-benefit balance of the product on the basis of scientific criteria concerning its quality, safety and efficacy.
Conditional Approval
In specific circumstances, E.U. legislation (Article 14(7) Regulation (EC) No 726/2004 and Regulation (EC) No 507/2006 on Conditional Marketing Authorizations for Medicinal Products for Human Use) enables applicants to obtain a conditional marketing authorization prior to obtaining the comprehensive clinical data required for an application for a full marketing authorization. Such conditional approvals may be granted for product candidates (including medicines designated as orphan medicinal products) if (i) the risk-benefit balance of the product candidate is positive, (ii) it is likely that the applicant will be in a position to provide the required comprehensive clinical trial data, (iii) the product fulfills unmet medical needs and (iv) the benefit to public health of the immediate availability on the market of the medicinal product concerned outweighs the risk inherent in the fact that additional data are still required. A conditional marketing authorization may contain specific obligations to be fulfilled by the marketing authorization holder, including obligations with respect to the completion of ongoing or new studies, and with respect to the collection of pharmacovigilance data. Conditional marketing authorizations are valid for one year, and may be renewed annually, if the risk-benefit balance remains positive, and after an assessment of the need for additional or modified conditions or specific obligations. The timelines for the centralized procedure described above also apply with respect to the review by the CHMP of applications for a conditional marketing authorization.
Pediatric Studies
Prior to obtaining a marketing authorization in the European Union, applicants have to demonstrate compliance with all measures included in an EMA-approved Pediatric Investigation Plan, or PIP, covering all subsets of the pediatric population, unless the EMA has granted a product-specific waiver, a class waiver, or a deferral for one or more of the measures included in the PIP. The respective requirements for all marketing authorization procedures are set forth in Regulation (EC) No 1901/2006, which is referred to as the Pediatric Regulation. This requirement also applies when a company wants to add a new indication, pharmaceutical form or route of administration for a medicine that is already authorized. The Pediatric Committee of the EMA, or PDCO, may grant deferrals for some medicines, allowing a company to delay development of the medicine in children until there is enough information to demonstrate its effectiveness and safety in adults. The PDCO may also grant waivers when development of a medicine in children is not needed or is not appropriate, such as for diseases that only affect the elderly population.
Before a marketing authorization application can be filed, or an existing marketing authorization can be amended, the EMA determines that companies actually comply with the agreed studies and measures listed in each relevant PIP.
European Union Regulatory Exclusivity
In the European Union, new products authorized for marketing (i.e., reference products) qualify for eight years of data exclusivity and an additional two years of market exclusivity upon marketing authorization. The data exclusivity period prevents generic or biosimilar applicants from relying on the preclinical and clinical trial data contained in the dossier of the reference product when applying for a generic or biosimilar marketing authorization in the European Union during a period of eight years from the date on which the reference product was first authorized in the European Union. The market exclusivity period prevents a successful generic or biosimilar applicant from commercializing its product in the EU until ten years have elapsed from the initial authorization of the reference product in the EU. The ten-year market exclusivity period can be extended to a maximum of eleven years if, during the first eight years of those ten years, the marketing authorization holder obtains an authorization for one or more new therapeutic indications which, during the scientific evaluation prior to their authorization, are held to bring a significant clinical benefit in comparison with existing therapies. In 2024, the European Parliament voted to adopt a new draft Regulation and draft Directive from the European Commission. These proposals set out significant amendments to the rules regarding regulatory data exclusivity and market protection for new medicines in Europe. The draft legislation will need to be approved by the European Council, before implementation in the EU. Once implemented, the Regulation will take effect in all EU Member States on a defined date, likely to be approximately in three to four years.
European Union Orphan Designation and Exclusivity
The criteria for designating an orphan medicinal product in the European Union, are similar in principle to those in the United States. Under Article 3 of Regulation (EC) 141/2000, a medicinal product may be designated as orphan if (i) it is intended for the diagnosis, prevention or treatment of a life-threatening or chronically debilitating condition; (ii) either (a) such condition affects no more than five in 10,000 persons in the European Union when the application is made, or (b) the product, without the benefits derived from orphan status, would not generate sufficient return in the European Union to justify investment; and (iii) there exists no satisfactory method of diagnosis, prevention or treatment of such condition authorized for marketing in the European Union, or if such a method exists, the product will be of significant benefit to those affected by the condition, as defined in Regulation (EC) 847/2000. Orphan medicinal products are eligible for financial incentives such as reduction of fees or fee waivers and are, upon grant of a marketing authorization, entitled to ten years of market exclusivity for the approved therapeutic indication. The application for orphan designation must be submitted before the application for marketing authorization. The applicant will receive a fee reduction for the marketing authorization application if the orphan designation has been granted, but not if the designation is still pending at the time the marketing authorization is submitted. Orphan designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process.
The ten-year market exclusivity in the European Union may be reduced to six years if, at the end of the fifth year, it is established that the product no longer meets the criteria for orphan designation, for example, if the product is sufficiently profitable not to justify maintenance of market exclusivity. Additionally, marketing authorization may be granted to a similar product for the same indication at any time if:
| • | the second applicant can establish that its product, although similar, is safer, more effective or otherwise clinically superior; |
| • | the applicant consents to a second orphan medicinal product application; or |
| • | the applicant cannot supply enough orphan medicinal product. |
PRIME Designation
The EMA grants access to the Priority Medicines, or PRIME, program to investigational medicines for which it determines there to be preliminary data available showing the potential to address an unmet medical need and bring a major therapeutic advantage to patients. As part of the program, EMA provides early and enhanced dialogue and support to optimize the development of eligible medicines and speed up their evaluation, aiming to bring promising treatments to patients sooner.
Periods of Authorization and Renewals
A marketing authorization is valid for five years in principle and the marketing authorization may be renewed after five years on the basis of a re-evaluation of the risk-benefit balance by the EMA or by the competent authority of the authorizing member state. To this end, the marketing authorization holder must provide the EMA or the competent authority with a consolidated version of the file in respect of quality, safety and efficacy, including all variations introduced since the marketing authorization was granted, at least six months before the marketing authorization ceases to be valid. Once renewed, the marketing authorization is valid for an unlimited period, unless the European Commission or the competent authority decides, on justified grounds relating to pharmacovigilance, to proceed with one additional five-year renewal. Any authorization which is not followed by the actual placing of the drug on the EU market (in case of centralized procedure) or on the market of the authorizing member state within three years after authorization ceases to be valid (the so-called sunset clause).
Rest of the World Regulation
For other countries outside of the European Union and the United States, such as countries in Eastern Europe, Latin America or Asia, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary from jurisdiction to jurisdiction. Additionally, the clinical trials must be conducted in accordance with GCP requirements and the applicable regulatory requirements and the ethical principles that have their origin in the Declaration of Helsinki.
If TuHURA fails to comply with applicable foreign regulatory requirements, TuHURA may be subject to, among other things, fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
Coverage, Pricing and Reimbursement
Sales of pharmaceutical products approved by the FDA will depend, in significant part, on the availability of third-party coverage and reimbursement for the products. Third-party payors include government healthcare programs in the United States such as Medicare and Medicaid, managed care providers, private health insurers and other organizations. These third-party payors are increasingly challenging the prices of products and examining the cost-effectiveness of medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare products. The process for determining whether a payor
will provide coverage for a product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the product once coverage is approved. Further, there is no uniform policy for coverage and reimbursement in the United States by third-party payors. Third-party payors may limit coverage to specific products on an approved list, or formulary, which might not include all of the approved products for a particular indication. TuHURA may need to conduct expensive pharmacoeconomic studies to demonstrate the medical necessity and cost-effectiveness of its products, in addition to the costs required to obtain FDA or other comparable regulatory approvals.
Moreover, a payor’s decision to provide coverage for a product does not imply that an adequate reimbursement rate will be approved. Third-party reimbursement may not be sufficient to maintain price levels high enough to realize an appropriate return on investment in product development. TuHURA’s product candidates may not be considered cost-effective. It is time consuming and expensive to seek coverage and reimbursement from third-party payors. Coverage and reimbursement may not be available or sufficient to allow TuHURA to sell its products on a competitive and profitable basis.
In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. Some countries provide that drug products may be marketed only after a reimbursement price has been agreed. Some countries may require the completion of additional studies that compare the cost-effectiveness of TuHURA’s product candidate to currently available therapies (so called health technology assessment, or HTA) in order to obtain reimbursement or pricing approval. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. An EU Member State may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the Company placing the medicinal product on the market. Other EU Member States allow companies to fix their own prices for drug products but monitor and control prescription volumes and issue guidance to physicians to limit prescriptions. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of TuHURA’s products. Historically, products launched in the European Union do not follow price structures of the United States and generally tend to be significantly lower.
The downward pressure on health care costs in general, particularly prescription drugs, has become intense. As a result, increasingly high barriers are being erected to the entry of new products. In addition, there can be considerable pressure by governments and other stakeholders on prices and reimbursement levels, including as part of cost containment measures. Political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after reimbursement has been obtained. Reference pricing used by various EU Member States and parallel distribution (arbitrage between low-priced and high- priced member states) can further reduce prices. Any country that has price controls or reimbursement limitations for drug products may not allow favorable reimbursement and pricing arrangements.
Other U.S. Health Care Laws and Regulations
Although TuHURA currently does not have any products on the market, TuHURA’s current and future arrangements with healthcare professionals, investigators, consultants, customers and third-party payors expose TuHURA to broadly applicable healthcare regulation and enforcement by the U.S. federal government and the states and foreign governments in which TuHURA conducts its business, such as fraud and abuse laws, transparency and health information privacy rules and regulations. These laws include, without limitation:
| • | The federal Anti-Kickback Statute – or AKS, 42 U.S.C. § 1320a-7b(b): the federal AKS is a criminal law which, prohibits, among other things, persons from knowingly and willfully soliciting, offering, receiving or paying remuneration, directly or indirectly, in cash or in kind, to induce or reward either the referral of an individual for the furnishing of any item or service, or for purchasing, leasing, |
| ordering, or arranging for or recommending purchasing, leasing, or ordering any good or service, for which payment may be made, in whole or in part, under a federal health care program such as Medicare and Medicaid. Remuneration includes anything of value and can take many forms besides cash, such as free rent, expensive hotel stays and meals, and excessive compensation for medical directorships or consultancies. The AKS covers the payers of kickbacks-those who offer or pay remuneration- as well as the recipients of kickbacks-those who solicit or receive remuneration. While each party’s intent is a key element of their liability under the AKS, a person or entity can be found guilty of violating the statute without actual knowledge of the statute or specific intent to violate it. A conviction for violation of the AKS can result in criminal fines and/or imprisonment and requires mandatory exclusion from participation in federal healthcare programs; |
| • | many US states have laws and regulations analogous to US federal fraud and abuse laws, such as individual state anti-kickback, fee-splitting and false claims laws, which may apply to sales or marketing arrangements and claims involving healthcare items or services reimbursed by non- governmental third-party payers; |
| • | The Federal civil and criminal false claims laws, including the civil False Claims Act, or the FCA,— 31 U.S.C. § § 3729-3733, which prohibits individuals or entities from knowingly presenting, or causing to be presented, to the federal government, claims for payment that are false or fraudulent or making a false statement to avoid, decrease or conceal an obligation to pay money to the federal government, and provides for civil whistleblower or qui tam actions that allow a private individual to file a lawsuit on behalf of the United State and entitles the whistleblower to a percentage of any recoveries. Under the FCA it is illegal to submit claims for payment to Medicare or Medicaid that an individual knows or should know are false or fraudulent; no specific intent to defraud is required. The civil FCA defines “knowing” to include not only actual knowledge but also instances in which the person acted in deliberate ignorance or reckless disregard of the truth or falsity of the information. Filing false claims may result in fines of up to three times the programs’ loss plus $11,000 per claim filed. Under the civil FCA, each instance of an item or a service billed to Medicare or Medicaid counts as a claim. The fact that a claim results from a kickback or is made in violation of the Stark law also may render it false or fraudulent, creating liability under the civil FCA as well as the AKS or Stark law. Under the criminal FCA (18 U.S.C. § 287) penalties for submitting false claims include imprisonment and criminal fines; the OIG also may impose administrative civil monetary penalties for false or fraudulent claims; |
| • | the federal civil monetary penalties law, or CMP (42 U.S.C. § 1320a-7a), prohibits a person from presenting or causing to be presented a claim that the provider knows or should know is improper, presenting a claim that the person knows or should know is for an item or service for which payment may not be made, and violating the AKS. The Office of Inspector General, or OIG of the US Department of Health and Human Services, or DHHS, may seek civil monetary penalties and sometimes exclusion for a wide variety of conduct and is authorized to seek different amounts of penalties and assessments based on the type of violation at issue; |
| • | the federal Health Insurance Portability and Accountability Act of 1996 and its implementing regulations, or HIPAA, imposes criminal and civil liability for executing a scheme to defraud any health care benefit program or making false statements relating to health care matters; |
| • | HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH Act, and its implementing regulations, also imposes obligations, including mandatory contractual terms, with respect to safeguarding the privacy, security and transmission of individually identifiable health information, for covered entities, including certain healthcare providers, health plans, and healthcare clearinghouses, and their business associates and covered subcontractors that provide services to, or on behalf of, the covered entity that involve individually identifiable health information; |
| • | The Physician Payments Sunshine Act (42 USC 1320a-7h) as known as “Open Payments” is a national disclosure program created by the Affordable Care Act, or ACA, that increases transparency into financial relationships between the health care industry (such as medical device manufacturers and pharmaceutical companies) and physicians or teaching hospitals. Drug, device, biological, and medical supply manufacturers, and group purchasing organizations are required to report payments or other transfers of value they make to physicians or teaching hospitals, as well as ownership or investment interests that a physician or his or her family members have in those entities. The Centers for Medicare & Medicaid Services, or CMS, collects data annually, and makes it publicly available and searchable online at openpaymentsdata.cms.gov. Applicable manufacturers are also required to report information related to payments and other transfers of value provided in the previous year to physician assistants, nurse practitioners, clinical nurse specialists, certified registered nurse anesthetists, and certified nurse midwives. Individual states have their own “sunshine act reporting laws” which vary from state to state; |
| • | the U.S. Foreign Corrupt Practices Act, or FCPA, and other anti-corruption laws and regulations pertaining to TuHURA’s financial relationships and interactions with foreign government officials, which prohibit U.S. companies and their employees, officers, and representatives from paying, offering to pay, promising, or authorizing the payment of anything of value to any foreign government official (including, potentially, healthcare professionals in countries in which TuHURA operates or may sell its products), government staff member, political party, or political candidate to obtain or retain business or to otherwise seek favorable treatment; |
| • | per the Exclusion Statute (42 U.S.C. § 1320a-7) the OIG is legally required to exclude from participation in all Federal health care programs individuals and entities convicted of the following types of criminal offenses: (1) Medicare or Medicaid fraud, as well as any other offenses related to the delivery of items or services under Medicare or Medicaid; (2) patient abuse or neglect; (3) felony convictions for other health-care-related fraud, theft, or other financial misconduct; and (4) felony convictions for unlawful manufacture, distribution, prescription, or dispensing of controlled substances. OIG has discretion to exclude individuals and entities on several other grounds, including misdemeanor convictions related to health care fraud other than Medicare or Medicaid fraud or misdemeanor convictions in connection with the unlawful manufacture, distribution, prescription, or dispensing of controlled substances; suspension, revocation, or surrender of a license to provide health care for reasons bearing on professional competence, professional performance, or financial integrity; provision of unnecessary or substandard services; submission of false or fraudulent claims to a Federal health care program; engaging in unlawful kickback arrangements; and defaulting on health education loan or scholarship obligations. If a person or entity is excluded by OIG from participation in the Federal health care programs, then Medicare, Medicaid, and other Federal health care programs, such as TRICARE and the Veterans Health Administration, will not pay for items or services that are furnished, ordered, or prescribed. Excluded physicians may not bill directly for treating Medicare and Medicaid patients, nor may their services be billed indirectly through an employer or a group practice. In addition, if you furnish services to a patient on a private-pay basis, no order or prescription that you give to that patient will be reimbursable by any Federal health care program; |
| • | the Physician Self-Referral Law, or the Stark Law - 42 U.S.C. § 1395nn, prohibits the submission, or causing the submission, of claims in violation of the law’s restrictions on referrals. The Stark Law prohibits a physician from referring Medicare patients to an entity (including pharmacies) for the furnishing of “designated health services,” if the physician or a member of the physician’s immediate family has a direct or indirect “financial relationship” with the entity, unless a specific exception applies. Financial relationships include both ownership/investment interests and compensation arrangements. The law further prohibits the entity from billing for any services that arise out of such prohibited referrals. Certain of these provisions are applicable to the referral of Medicaid patients as well. Designated health services include outpatient prescription drug services; clinical laboratory services; physical therapy, occupational therapy, and outpatient speech-language pathology services; |
| radiology and certain other imaging services; radiation therapy services and supplies; DME and supplies; parenteral and enteral nutrients, equipment, and supplies; prosthetics, orthotics, and prosthetic devices and supplies; home health services; and inpatient and outpatient hospital services. The Stark Law is a strict liability statute thus the prohibition applies regardless of the rationale for the financial relationship and the reason for ordering the service; and |
| • | analogous state and foreign laws and regulations, such as state anti-kickback and false claims laws, may apply to sales or marketing arrangements and claims involving health care items or services reimbursed by nongovernmental third-party payors, including private insurers. |
Some state laws require pharmaceutical companies to comply with the pharmaceutical industry’s voluntary compliance guidelines, such as the PhRMA Code, or the relevant compliance guidance promulgated by the federal government, in addition to requiring drug manufacturers to report information related to payments to physicians and other health care providers or marketing expenditures to the extent that those laws impose requirements that are more stringent than the Physician Payments Sunshine Act. In addition, state and local laws may require the registration of pharmaceutical sales representatives. State and foreign laws also govern the privacy and security of health information in some circumstances, many of which differ from each other in significant ways and often are not preempted by HIPAA, thus complicating compliance efforts.
Violations of any of such laws or any other governmental regulations that apply to us, may subject TuHURA to significant penalties, including, without limitation, civil, criminal and administrative penalties, damages, fines, disgorgement, additional reporting requirements and oversight if the Company becomes subject to a corporate integrity agreement or similar agreement to resolve allegations of non-compliance with these laws, the curtailment or restructuring of TuHURA’s operations, exclusion from participation in federal and state healthcare programs and imprisonment, any of which could adversely affect TuHURA’s ability to operate its business.
Health Care Reform in the United States and Potential Changes to Health Care Laws
The FDA’s and other regulatory authorities’ policies may change and additional government regulations may be enacted that could prevent, limit or delay regulatory approval of TuHURA’s product candidates. If TuHURA is slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if TuHURA is not able to maintain regulatory compliance, TuHURA may lose any marketing approval that TuHURA otherwise may have obtained and may not achieve or sustain profitability, which would adversely affect its business, prospects, financial condition and results of operations.
As previously mentioned, a primary trend in the U.S. health care industry and elsewhere is cost containment. Government authorities and other third-party payors have attempted to control costs by limiting coverage and the amount of reimbursement for particular medical products and services, implementing reductions in Medicare and other health care funding and applying new payment methodologies. For example, in March 2010, the ACA was enacted, which, among other things, increased the minimum Medicaid rebates owed by most manufacturers under the Medicaid Drug Rebate Program; introduced a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; extended the Medicaid Drug Rebate Program to utilization of prescriptions of individuals enrolled in Medicaid managed care plans; imposed mandatory discounts for certain Medicare Part D beneficiaries as a condition for manufacturers’ outpatient drugs coverage under Medicare Part D; and established a Center for Medicare Innovation at CMS to test innovative payment and service delivery models to lower Medicare and Medicaid spending.
In addition, other legislative changes have been proposed and adopted in the United States since the ACA that affect health care expenditures. There has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries, presidential executive orders and proposed and enacted federal and state legislation designed to, among other
things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs and reform government program reimbursement methodologies for pharmaceutical and biologic products. Notably, on December 20, 2019, President Trump signed the Further Consolidated Appropriations Act for 2020 into law (P.L. 116-94) that includes a piece of bipartisan legislation called the Creating and Restoring Equal Access to Equivalent Samples Act of 2019 or the “CREATES Act.” The CREATES Act aims to address the concern articulated by both the FDA and others in the industry that some brand manufacturers have improperly restricted the distribution of their products, including by invoking the existence of a REMS for certain products, to deny generic and biosimilar product developers access to samples of brand products. Because generic and biosimilar product developers need samples to conduct certain comparative testing required by the FDA, some have attributed the inability to timely obtain samples as a cause of delay in the entry of generic and biosimilar products. To remedy this concern, the CREATES Act establishes a private cause of action that permits a generic or biosimilar product developer to sue the brand manufacturer to compel it to furnish the necessary samples on “commercially reasonable, market-based terms.” Whether and how generic and biosimilar product developments will use this new pathway, as well as the likely outcome of any legal challenges to provisions of the CREATES Act, remain highly uncertain and its potential effects on TuHURA’s future commercial products are unknown. The FDA also released a final rule on September 24, 2020 providing guidance for states to build and submit importation plans for drugs from Canada. This final rule became effective November 30, 2020. In January 2024, FDA authorized the state of Florida’s Section 804 Importance Program to allow Florida to import drugs from Canada for a period of two years. The ongoing impact of this and potentially other state programs is still unclear.
TuHURA cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative or executive action, either in the United States or abroad. TuHURA expects that additional state and federal health care reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for health care products and services.